The SignalP 5.0 server predicts the presence of signal peptides and the location of their cleavage sites in proteins from Archaea, Gram-positive Bacteria, Gram-negative Bacteria and Eukarya. In Bacteria and Archaea, SignalP 5.0 can discriminate between three types of signal peptides:
SignalP 5.0 is based on a deep convolutional and recurrent neural network architecture including a conditional random field.
Click here to read "A Brief History of Protein Sorting Prediction", The Protein Journal, 2019
Remember, the presence or absence of a signal peptide is not the whole story about the localization of a protein! If you want to find out more about the sorting of your eukaryotic proteins, try the protein subcellular localization predictor.
Instructions
1. Specify the input sequences
All the input sequences must be in one-letter amino acid
code. The allowed alphabet (not case sensitive) is as follows:
A C D E F G H I K L M N P Q R S T V W Y and X (unknown)
All the alphabetic symbols not in the allowed alphabet
will be converted to X before processing. All the non-alphabetic
symbols, including white space and digits, will be ignored.
The sequences can be input in the following two ways:
-
Paste a single sequence (just the amino acids) or a number of sequences in
FASTA
format into the upper window of the main server page.
-
Select a FASTA
file on your local disk, either by typing the file name into the lower window
or by browsing the disk.
Both ways can be employed at the same time: all the specified sequences will
be processed. However, there may be not more than 5,000 sequences in one submission. The sequences
may not be longer than 10,000 amino acids.
2. Customize your run
- Organism group:
It is important for performance that you choose the correct organism
group —
Archaea, Eukaryotes, Gram-negative bacteria or Gram-positive bacteria —
since the signal peptides of these three groups are known to differ
from each other.
Gram-positive bacteria correspond to
Actinobacteria and
Firmicutes in the
NCBI Taxonomy.
Gram-negative bacteria are all other
eubacteria, except
Tenericutes (including
Mycoplasma), which seem to lack a type I signal peptidase and
therefore do not have standard signal peptides.
- Output format:
You can choose between four output formats:
- Standard
- Appropriate for most users. Shows one plot and one summary per sequence.
- Short
- Convenient if you submit lots of sequences. Shows only one line of
output per sequence and no graphics.
3. Submit the job
Click on the
"Submit" button. The status of your job (either 'queued'
or 'running') will be displayed and constantly updated until it terminates and
the server output appears in the browser window.
At any time during the wait you may enter your e-mail address and simply leave
the window. Your job will continue; you will be notified by e-mail when it has
terminated. The e-mail message will contain the URL under which the results are
stored; they will remain on the server for 24 hours for you to collect them.
Example Outputs
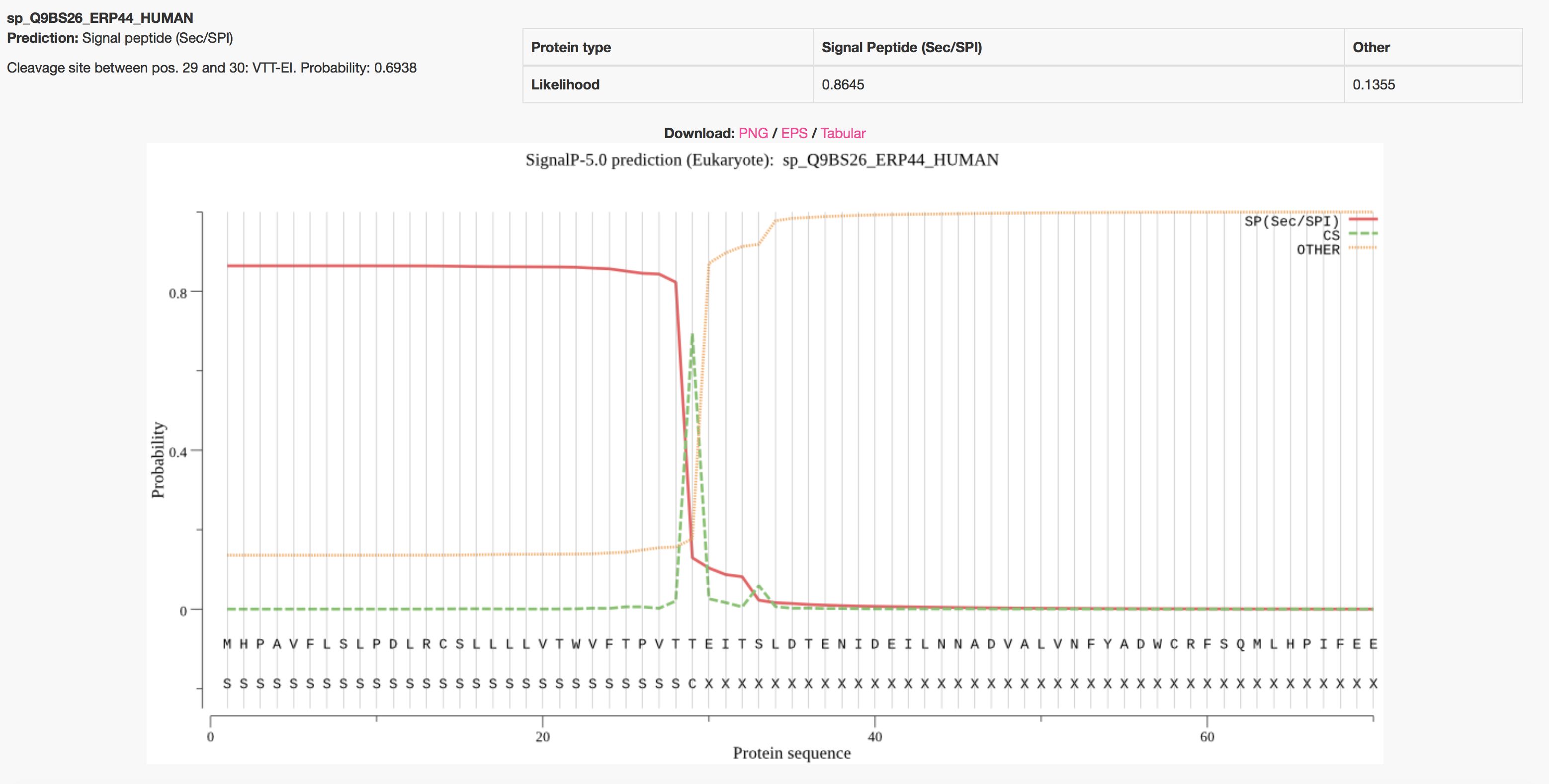
By default the server produces the following output for each input sequence. The example below shows the output for thioredoxin domain containing protein 4 precursor (endoplasmic reticulum protein ERp44), taken from the
Uniprot entry
ERP44_HUMAN. The signal peptide prediction is consistent with the database annotation.
One annotation is attributed to each protein, the one that has the highest probability. The protein can have a Sec signal peptide (Sec/SPI), a Lipoprotein signal peptide (Sec/SPII), a Tat signal peptide (Tat/SPI) or No signal peptide at all (Other).
If a signal peptide is predicted, the cleavage site position is reported as well.
On the plot, three marginal probabilities are reported, i.e. SP(Sec/SPI) / LIPO(Sec/SPII) / TAT(Tat/SPI) (depending on what type of signal peptide is predicted), CS (the cleavage site) and OTHER (the probability that the sequence does not have any kind of signal peptide).
Example: secretory protein - standard output format
Example: secretory protein - short output format

From the Downloads tab, the user can obtain the results of the run in various formats, i.e.
JSON, Prediction summary (results for each submission, 1 line per sequence), Processed entries fasta (a FASTA sequence file containing the sequences of protein that had predicted signal peptides, with the signal peptide removed) and Processed entries
gff3 (a file showing the signal peptides feature of those proteins that had predicted signal peptides in GFF3 format).
Training and testing data sets
The datasets for training and testing SignalP5.0 can be found here. Both datasets are in 3-line FASTA format:
>Uniprot_AC|Kingdom|Type|Partition No
amino-acid sequence
annotation [S: Sec/SPI signal peptide | T: Tat/SPI signal peptide | L: Sec/SPII signal peptide | I: cytoplasm | M: transmembrane | O: extracellular]
Training set: download
Benchmark set: download
Article abstracts
Main references:
Other publications
Henrik Nielsen's PhD thesis
Current version (SignalP v. 5.0)
SignalP 5.0 improves signal peptide predictions using deep neural networks.
José Juan Almagro Armenteros, Konstantinos D. Tsirigos, Casper Kaae Sønderby, Thomas Nordahl Petersen, Ole Winther, Søren Brunak, Gunnar von Heijne and Henrik Nielsen.
Nature Biotechnology,
37, 420-423, doi:
10.1038/s41587-019-0036-z (2019)
Signal peptides (SPs) are short amino acid sequences in the amino terminus of many newly synthesized proteins that target proteins into, or across, membranes. Bioinformatic tools can predict SPs from amino acid sequences, but most cannot distinguish between various types of signal peptides. We present a deep neural network-based approach that improves SP prediction across all domains of life and distinguishes between three types of prokaryotic SPs.
PMID: 30778233
Original method (SignalP v. 1.1)
Identification of prokaryotic and eukaryotic signal peptides
and prediction of their cleavage sites.
Henrik Nielsen, Jacob Engelbrecht, Søren Brunak and Gunnar von
Heijne.
Protein Engineering,
10:1-6 (1997).
We have developed a new method for the identification of signal peptides and
their cleavage sites based on neural networks trained on separate sets of
prokaryotic and eukaryotic sequence. The method performs significantly better
than previous prediction schemes and can easily be applied on genome-wide data
sets. Discrimination between cleaved signal peptides and uncleaved N-terminal
signal-anchor sequences is also possible, though with lower precision.
Predictions can be made on a publicly available WWW server.
PMID: 9051728
(free full text pdf
version)
Update to SignalP v. 2.0
Prediction of signal peptides and signal anchors by a hidden Markov
model.
Henrik Nielsen and Anders Krogh.
Proc Int Conf Intell Syst Mol Biol. (ISMB 6),
6:122-130 (1998).
A hidden Markov model of signal peptides has been developed. It contains
submodels for the N-terminal part, the hydrophobic region, and the region
around the cleavage site. For known signal peptides, the model can be used to
assign objective boundaries between these three regions. Applied to our data,
the length distributions for the three regions are significantly different from
expectations. For instance, the assigned hydrophobic region is between 8 and 12
residues long in almost all eukaryotic signal peptides. This analysis also
makes obvious the difference between eukaryotes, Gram-positive bacteria, and
Gram-negative bacteria. The model can be used to predict the location of the
cleavage site, which it finds correctly in nearly 70% of signal peptides in a
cross-validated test — almost the same accuracy as the best previous method. One
of the problems for existing prediction methods is the poor discrimination
between signal peptides and uncleaved signal anchors, but this is substantially
improved by the hidden Markov model when expanding it with a very simple signal
anchor model.
PMID: 9783217
Update to SignalP v. 3.0
Improved prediction of signal peptides: SignalP 3.0.
Jannick Dyrløv Bendtsen, Henrik Nielsen,
Gunnar von Heijne and Søren Brunak.
J. Mol. Biol.,
340:783-795 (2004).
We describe improvements of the currently most
popular method for prediction of classically secreted proteins,
SignalP. SignalP consists of two different predictors based on
neural network and hidden Markov model algorithms, and both
components have been updated. Motivated by the idea that the
cleavage site position and the amino acid composition of the
signal peptide are correlated, new features have been included as
input to the neural network. This addition, together with a
thorough error-correction of a new data set, have improved the
performance of the predictor significantly over SignalP version 2.
In version 3, correctness of the cleavage site predictions have
increased notably for all three organism groups, eukaryotes, Gram
negative and Gram positive bacteria. The accuracy of cleavage site
prediction has increased in the range from 6–17 % over the
previous version, whereas the signal peptide discrimination
improvement mainly is due to the elimination of false positive
predictions, as well as the introduction of a new discrimination
score for the neural network. The new method has also been
benchmarked against other available methods.
PMID: 15223320
doi: 10.1016/j.jmb.2004.05.028
Update to SignalP v. 4.0
SignalP 4.0: discriminating signal peptides from transmembrane regions.
Thomas Nordahl Petersen, Søren Brunak,
Gunnar von Heijne and Henrik Nielsen.
Nature Methods,
8:785-786 (2011).
This is a Correspondence, it has no abstract.
doi: 10.1038/nmeth.1701
Access to the supplementary materials:
nmeth.1701-S1.pdf
Update to SignalP v. 4.1
Predicting Secretory Proteins with SignalP
Henrik Nielsen.
In Kihara, D (ed):
Protein Function Prediction (Methods in Molecular Biology vol. 1611) pp. 59-73, Springer 2017.
doi:
10.1007/978-1-4939-7015-5_6
PMID:
28451972
SignalP is the currently most widely used program for prediction of signal peptides from amino acid sequences. Proteins with signal peptides are targeted to the secretory pathway, but are not necessarily secreted. After a brief introduction to the biology of signal peptides and the history of signal peptide prediction, this chapter will describe all the options of the current version of SignalP and the details of the output from the program. The chapter includes a case study where the scores of SignalP were used in a novel way to predict the functional effects of amino acid substitutions in signal peptides.
Other publications
Locating proteins in the cell using TargetP,
SignalP, and related tools
Olof Emanuelsson, Søren Brunak, Gunnar von Heijne, Henrik Nielsen
Nature Protocols, 2:953-971 (2007).
Determining the subcellular localization of a protein is an important
first step toward understanding its function. Here, we describe the
properties of three well-known N-terminal sequence motifs directing
proteins to the secretory pathway, mitochondria and chloroplasts, and
sketch a brief history of methods to predict subcellular localization
based on these sorting signals and other sequence properties. We then
outline how to use a number of internet-accessible tools to arrive at a
reliable subcellular localization prediction for eukaryotic and
prokaryotic proteins. In particular, we provide detailed step-by-step
instructions for the coupled use of the amino-acid sequence-based
predictors TargetP, SignalP, ChloroP and TMHMM, which are all hosted at
the Center for Biological Sequence Analysis, Technical University of
Denmark. In addition, we describe and provide web references to other
useful subcellular localization predictors. Finally, we discuss
predictive performance measures in general and the performance of
TargetP and SignalP in particular.
PMID: 17446895
Please click
here to access the
paper and supplementary materials.
Machine learning approaches to the prediction of signal peptides
and other protein sorting signals.
Henrik Nielsen, Søren Brunak, and Gunnar von Heijne.
Protein Engineering, 12:3-9 (1999), Review.
Prediction of protein sorting signals from the sequence of amino acids has
great importance in the field of proteomics today. Recently, the growth of
protein databases, combined with machine learning approaches, such as neural
networks and hidden Markov models, have made it possible to achieve a level of
reliability where practical use in, for example automatic database annotation
is feasible. In this review, we concentrate on the present status and future
perspectives of SignalP, our neural network-based method for prediction of the
most well-known sorting signal: the secretory signal peptide. We discuss the
problems associated with the use of SignalP on genomic sequences, showing that
signal peptide prediction will improve further if integrated with predictions
of start codons and transmembrane helices. As a step towards this goal, a
hidden Markov model version of SignalP has been developed, making it possible
to discriminate between cleaved signal peptides and uncleaved signal anchors.
Furthermore, we show how SignalP can be used to characterize putative signal
peptides from an archaeon, Methanococcus jannaschii. Finally, we briefly review
a few methods for predicting other protein sorting signals and discuss the
future of protein sorting prediction in general.
PMID: 10065704
A neural network method for identification of prokaryotic and eukaryotic
signal peptides and prediction of their cleavage sites.
Henrik Nielsen, Jacob Engelbrecht, Søren Brunak
and Gunnar von Heijne.
Int. J. Neural Sys., 8:581-599 (1997).
We have developed a new method for the identification of signal peptides and
their cleavage sites based on neural networks trained on separate sets of
prokaryotic and eukaryotic sequences. The method performs significantly better
than previous prediction schemes, and can easily be applied to genome-wide data
sets. Discrimination between cleaved signal peptides and uncleaved N-terminal
signal-anchor sequences is also possible, though with lower precision.
Predictions can be made on a publicly available WWW server:
http://www.cbs.dtu.dk/services/SignalP/.
PMID: 10065837
Defining a similarity threshold for a functional protein sequence pattern:
the signal peptide cleavage site.
Henrik Nielsen, Jacob Engelbrecht, Gunnar von Heijne
and Søren Brunak.
Proteins, 24:165-77 (1996).
When preparing data sets of amino acid or nucleotide sequences it is
necessary to exclude redundant or homologous sequences in order to avoid
overestimating the predictive performance of an algorithm. For some time
methods for doing this have been available in the area of protein structure
prediction. We have developed a similar procedure based on pair-wise
alignments for sequences with functional sites. We show how a correlation
coefficient between sequence similarity and functional homology can be used
to compare the efficiency of different similarity measures and choose a
nonarbitrary threshold value for excluding redundant sequences. The impact
of the choice of scoring matrix used in the alignments is examined. We
demonstrate that the parameter determining the quality of the correlation is
the relative entropy of the matrix, rather than the assumed (PAM or
identity) substitution mode. Results are presented for the case of
prediction of cleavage sites in signal peptides. By inspection of the false
positives, several errors in the database were found. The procedure
presented may be used as a general outline for finding a problem-specific
similarity measure and threshold value for analysis of other functional
amino acid or nucleotide sequence patterns.
PMID: 8820484
From sequence to sorting: Prediction of signal peptides.
Henrik Nielsen.
Ph.D. thesis, defended at Department of Biochemistry,
Stockholm University, Sweden, May 25, 1999.
In the present age of genome sequencing, a vast number of predicted
genes are initially known only by their putative nucleotide
sequence. The newly established field of bioinformatics is concerned
with the computational prediction of structural and functional
properties of genes and the proteins they encode, based on their
nucleotide and amino acid sequences.
Since one of the crucial properties of a protein is its subcellular
location, prediction of protein sorting is an important question in
bioinformatics. A fundamental distinction in protein sorting is that
between secretory and non-secretory proteins, determined by a
cleavable N-terminal sorting signal, the secretory signal peptide.
The main part of this thesis, including four of the six papers,
concerns prediction of secretory signal peptides in both eukaryotic
and bacterial data using two machine learning techniques: artificial
neural networks and hidden Markov models. A central result is the
SignalP prediction method, which has been made available as a World
Wide Web server and is very widely used.
Two additional prediction methods are also included, with one paper
each. ChloroP predicts chloroplast transit peptides, another
cleavable N-terminal sorting signal; while NetStart predicts start
codons in eukaryotic genes. For prediction of all N-terminal signals,
the assignment of correct start codon can be critical, which is why
prediction of translation initiation from the nucleotide sequence is
also important for protein sorting prediction.
This thesis comprises a detailed review of the molecular biology of
protein secretion, a short introduction to the most important machine
learning algorithms in bioinformatics, and a critical review of
existing methods for protein sorting prediction. In addition, it
contains general treatment of the principles of data set construction
and performance evaluation for prediction methods in bioinformatics.
Access to the thesis (without the six included papers):
PhDthesis.pdf; PhDthesis-cover.pdf
Frequently Asked Questions
Changes from version 4 to 5
Changes from version 4.0 to 4.1
Changes from version 3 to 4
Biological background, signal peptides
Biological background, other sorting signals
Biological background, organism groups
History
— What's new?
Please see the version history page.
— What happened to the C-, S- and Y-scores?
The output layer of SignalP 5.0 is a conditional random field (CRF) which
yields marginal probabilities, just like the HMM module did in SignalP versions
2 and 3. Since the CRF is a grammatical method which is aware that there can
only be one cleavage site in a given signal peptide, there is no need for
the post-processing of the network output that was represented by the Y-score.
— What's new?
Please see the version history page.
— Why do you present a choice between two cutoff settings?
Can't you just decide on one?
The optimal cutoff really depends on what you want to use the method
for. If it is important to find all signal peptides, use the sensitive
cutoff. If you want an estimate of the number of signal peptides in a
genome, use the default cutoff.
— Why have you imposed a minimum length?
Because we believe that predictions of signal peptides
shorter than ten residues made by SignalP 4.1 are false. The shortest
known signal peptides are 11 residues long (with one exception,
SP23_TENMO,
which does not look like a signal peptide at
all). Click
here for an updated list of experimentally confirmed signal
peptides from UniProt of length 11 or shorter.
— What happened to the Background page?
It's here! The important material from the Background page has been
integrated into this FAQ, we hope you like the new format.
— What's new?
Please see the version history page.
— What happened to the HMM part?
While making SignalP 4.0, we did retrain the Hidden Markov Model (HMM)
part of SignalP. However, we found that it did not perform better than
the neural networks in any of the performance parameters we tested.
Therefore, we decided not to include it. If the HMM output is important
for you, you can still use
SignalP 3.0.
— Why is my favourite signal peptide no longer predicted correctly?
SignalP 3.0 could do it!
As explained on the performance page,
SignalP 4 with the default cutoff has a lower sensitivity than SignalP
3. Please try again with the new "Sensitive" setting.
— What happened to the Yes/No answers for max C score etc.?
SignalP 3.0 provided five Yes/No answers for the NN part. We found that
this was confusing for users and obscured the fact that the D-score is
the best score for discriminating between signal peptides and non-signal
peptides.
— What are signal peptides?
The term "signal peptide" is used with two meanings: In the broad
sense (used in many textbooks),
a signal peptide is any sorting signal embedded in the amino
acid sequence of a protein. In the narrow sense (used in most of
the scientific literature), a signal peptide
is an N-terminal signal that directs the protein across the ER
membrane in eukaryotes and across the plasma membrane in prokaryotes.
Signal peptides in the narrow sense are also known as ER signal
peptides or secretory signal peptides. Read more in
UniProt, in
Wikipedia,
and in the
Sequence feature ontology.
It is important to emphasize that SignalP predicts signal peptides in
the narrow sense only.
— Are signal peptides always N-terminal?
In the narrow sense: Yes, per definition. In the broad sense: No,
there are several sorting signal that are C-terminal
(e.g. the PTS1 signal for peroxisomal import)
or internal (e.g. the nuclear localization signal).
— Are signal peptides (in the narrow sense) always cleaved?
No, there are rare cases of uncleaved signal peptides. For an updated
list of such proteins annotated in UniProt, click
here.
These should not be confused with signal anchors, see below.
— Which protease is responsible for signal peptide (Sec/SPI)
cleavage?
In bacteria, it is Signal Peptidase I (SPase I), also known as Leader Peptidase
(
Lep). In eukaryotes, it is the signal peptidase complex (SPC), which
consists of four subunits in yeast and five in mammals.
Read more in
MEROPS.
— My protein has a signal peptide. Can I then safely
conclude that it is secreted?
No. You can only conclude that it enters the secretory pathway.
In eukaryotes, there are several opportunities for a protein with a
signal peptide to escape secretion. It could:
- be retained in the endoplasmic reticulum (ER). Soluble ER-resident
proteins have a C-terminal retention signal with the consensus
sequence KDEL, see
PROSITE.
- be retained in the Golgi apparatus,
- be directed to the lysosome (vacuole in plants and fungi),
- have one or more transmembrane helices and therefore be
retained in either the plasma membrane, or one of the membranes of the
secretory pathway (ER, Golgi, lysosome/vacuole), or
- have a signal for GPI-anchoring, a C-terminal cleaved
peptide which functions as a signal for attachment of a
Glycophosphatidylinositol
(GPI) group that anchors the protein to the
outer face of the plasma membrane.
In
Gram-positive bacteria and
Archaea, a protein with a signal peptide could:
- have one or more transmembrane helices, or
- be attached to the cell wall.
In Gram-negative bacteria, a protein with a signal peptide could:
- have one or more transmembrane helices,
- be retained in the periplasm, or
- be inserted into the outer membrane as a β-barrel transmembrane
protein.
— Does SignalP predict signal peptides of bacterial and archaeal
lipoproteins?
Yes. Bacterial lipoproteins have special signal peptides (Sec/SPII) which are
cleaved by Signal Peptidase II (SPase II), also known as Lipoprotein
signal peptidase (
Lsp). A diacylglyceryl group is attached to a Cysteine residue
in position +1 relative to the cleavage site, which bears no resemblance
to the SPase I cleavage site. See also
MEROPS
and
PROSITE.
— Does SignalP predict Tat (Twin-arginine translocation) signal peptides?
Yes. Bacterial and archaeal Tat signal peptides (Tat/SPI), which direct their proteins through
an alternative translocon (
TatABC instead of
SecYEG),
have a special motif, usually containing two
Arginines, in the n-region. Additionally, they are in general longer and less
hydrophobic than "normal" (
Sec) signal peptides. See also
PROSITE and
InterPro.
— What about Tat/SPII signal peptides?
SignalP cannot predict lipoprotein signal peptides that are transported through
the Tat translocon and cleaved by SPase II. When constructing the datasets
for SignalP 5.0, we gathered Tat signal peptides from
PROSITE PS51318 and
PRED-TAT,
while lipoprotein signal peptides were gathered from
PROSITE PS51257 and
PRED-LIPO, and there was
no overlap between them. even though such proteins are known to exist
(
Thompson et al. 2010).
— What are signal anchors?
A signal anchor is a transmembrane helix located close to the N-terminus
of a protein with an N-in orientation (i.e. the N-terminus is on the
cytoplasmic side of the membrane). It functions much like a signal
peptide since it is recognized by the Signal Recognition Particle (SRP)
and inserted into the translocon; but instead of being cleaved and
degraded it remains in the membrane and anchors the protein to it.
Proteins anchored in this way are known as Type II transmembrane
proteins.
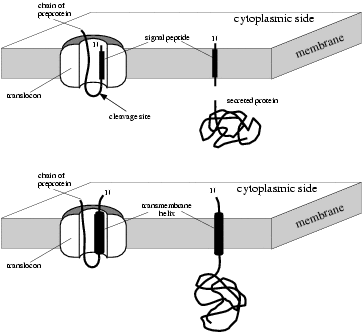 |
Signal peptides (above) versus
signal anchors (below) |
It is important to realize that the difference between signal peptides
and signal anchors is not a question of presence or absence of a
cleavage site. Instead, the most important difference seems to be the
length of the hydrophobic domain. It has been shown experimentally that
it is possible to convert a cleaved
signal peptide to a signal anchor merely by lengthening the
h-region, without altering the cleavage site
(
Chou & Kendall 1990;
Nilsson, Whitley, & von Heijne 1994).
The introduction of the Hidden Markov Model (HMM) method in SignalP
version 2 made it possible to some extent to distinguish signal peptides
from signal anchors (in that version, only in eukaryotes). However,
SignalP 4 (based entirely on the Neural Network (NN) method), does a
better job, since its negative set is not confined only to transmembrane
helices annotated as signal anchors, but includes all types of
transmembrane segments close to the N-terminus.
— What should I use for predicting signal peptides in the
broad sense?
For mitochondrial and plastid import signals, also known as
transit
peptides, we recommend
TargetP. For
general prediction of subcellular location in eukaryotes, we recommend
DeepLoc.
— What should I use for predicting non-classical
(leaderless) secreted proteins?
Not all secretory proteins carry signal peptides. Some proteins enter a non-classical secretory pathway
without any currently known sequence motif. In eukaryotes, these proteins are mostly growth factors
and extracellular matrix binding proteins. In Gram-negative bacteria, the
type I, III, IV and VI secretion systems function without signal peptides.
For prediction of such proteins we
recommend the
SecretomeP
server.
— Which version should I use for vira and bacteriophages?
You should use the version corresponding to the host organism. There are
some indications that viral signal peptides differ from those of the
host organism, but SignalP currently does not take that into account.
— Which version should I use for Tenericutes/Mollicutes
(Mycoplasma and related genera)?
You shouldn't use SignalP at all for these organisms, since they seem to
lack a type I signal peptidase completely!
— Which version should I use for metagenomic sequences
of unknown origin?
This is an unsolved question. Please use all four versions to
search for signal peptides in such data.
— Is one version enough for all eukaryotic organisms, or
are there differences within the eukaryotes?
It is known that some yeast signal peptides are not recognized by
mammalian cells (Bird
et al.,
1987 and
1990).
Therefore, it would be natural to assume that separate SignalP versions
for yeast and Mammalia would provide better predictions than a common
eukaryotic version. While developing SignalP 4.0 we tried dividing the
eukaryotic data into animals, fungi, and plants and training separate
methods for these three groups. However, this did not give any
improvement, and performance for all three groups was better when using
the method trained on all eukaryotic sequences together.
— Are two versions enough for all bacteria, or
are there differences within the Gram-positive/Gram-negative
bacterial groups?
The Gram-negative version of SignalP is almost certainly biased towards
E. coli and other
γ-proteobacteria,
since these constitute the bulk
of the experimentally annotated bacterial proteins in UniProt.
Unpublished results suggest that some bacteria have very divergent
cleavage site motifs. Future versions of SignalP might therefore divide
the Gram-negative bacteria into several classes, if data are available.
Gram-positive bacteria probably constitute a more homogenous group, but
it is an open question whether there are differences in signal peptides
between
Actinobacteria (high G+C Gram-positive bacteria) and
Firmicutes (low G+C Gram-positive bacteria). More data on
Actinobacteria are needed before that can be answered.
— How are the various versions of SignalP related?
Please see the version history page
— Was there ever a Nobel prize awarded for signal peptides?
Yes, for signal peptides in the broad sense. The importance of signal peptides
was emphasized in 1999 when Günter Blobel received the Nobel Prize in
physiology or medicine for his discovery "proteins have intrinsic
signal that govern their transport and localization in the cell".
See
the
press release.
— Was SignalP the first signal peptide predictor?
No, but it was, to our knowledge, the first to be implemented as a
web server (in 1996). Among the earlier methods were
McGeoch (1985)
and
von Heijne (1986),
both of which have been included in
PSORT.
— How many times have the SignalP papers been cited?
This information is available on Henrik Nielsen's
ResearcherID,
Scopus,
and
Google
Scholar pages.
Version history
Please click on the version number to activate the corresponding server where available.
|
5.0
|
The current server. New in this version:
- Deep learning:
SignalP 5.0 is based on convolutional and recurrent (LSTM) neural
networks.
The deep recurrent neural network architecture is better suited to
recognizing sequence motifs of varying length, such as signal peptides, than
traditional feed-forward neural networks (as used in SignalP 1-4).
- Conditional random field:
The neural networks of SignalP 5.0 are
combined with a conditional random field (CRF). The CRF
imposes a defined grammar on the prediction and obviates the need for
the post-processing step (Y- and D-scores) used in earlier versions of
SignalP.
- Transfer learning:
Instead of training separate networks for each organism group, SignalP 5.0
exploits the fact that signal peptides from different domains of life are
to some degree similar. Thus, SignalP 5.0 is trained on data from all groups
while an extra input unit informs the network about the origin of the
sequences.
- Archaeal option:
Thanks to transfer learning, SignalP 5.0 is able to make predictions also of
signal peptides from Archaea, even though the data set is limited.
- Lipoprotein signal peptides:
SignalP 5.0 can now differentiate between "standard" signal peptidase
I-cleaved signal peptides (Sec/SPI) and signal peptidase II-cleaved
lipoprotein signal peptides (Sec/SPII) in Bacteria and Archaea.
Previously, we referred to the LipoP
server for this prediction.
- Tat signal peptides:
SignalP 5.0 can now differentiate between "standard" signal peptides translocated by
the Sec translocon (Sec/SPI) and
"Tat" (Twin-Arginine Translocation) signal peptides
translocated by the Tat translocon (Tat/SPI) in Bacteria and Archaea.
Previously, we referred to the TatP
server for this prediction. However, SignalP 5.0 cannot predict
lipoprotein signal peptides translocated by the Tat translocon
(Tat/SPII) since we did not find any confirmed examples of these while
constructing the data sets.
Main publication:
-
SignalP 5.0 improves signal peptide predictions using deep neural networks
José Juan Almagro Armenteros, Konstantinos D. Tsirigos,
Casper Kaae Sønderby, Thomas Nordahl Petersen, Ole Winther,
Søren Brunak,
Gunnar von Heijne and Henrik Nielsen.
Nature Biotechnology, , 2019.
|
|
4.1
|
The current server. New in this version:
- For the web page, an option to set the D-score cutoff values so
that the sensitivity is the same as that of SignalP 3.0.
- Option included to set the minimum cleavage site position i.e. Ymax position - default value is 10.
- For the signalp package an option has been included to specify a temporary directory (-T dir).
- For the signalp package an option has been included to show signalp version (-V).
- Documentation rewritten.
Main publication:
-
SignalP 4.0: discriminating signal peptides from transmembrane regions
Thomas Nordahl Petersen, Søren Brunak,
Gunnar von Heijne and Henrik Nielsen.
Nature Methods, 8:785-786, 2011.
|
|
4.0
|
New in this version:
- Improved discrimination between signal peptides and transmembrane regions.
- No HMM method - only one prediction.
Main publication:
-
SignalP 4.0: discriminating signal peptides from transmembrane regions
Thomas Nordahl Petersen, Søren Brunak,
Gunnar von Heijne and Henrik Nielsen.
Nature Methods, 8:785-786, 2011.
|
|
3.0
|
New in this version:
- D-score. Improved quality of prediction.
Main publication:
-
Improved prediction of signal peptides: SignalP 3.0.
Jannick Dyrløv Bendtsen, Henrik Nielsen,
Gunnar von Heijne and Søren Brunak.
J. Mol. Biol., 340:783-795, 2004.
|
|
2.0
|
New in this version:
- Incorporation of a hidden Markov model version:
SignalP V2.0 comprises two signal peptide prediction methods,
SignalP-NN (based on neural networks, corresponding to SignalP V1.1)
and SignalP-HMM (based on hidden Markov models). For eukaryotic data,
SignalP-HMM has a substantially improved discrimination between signal
peptides and uncleaved signal anchors, but it has a slightly lower
accuracy in predicting the precise location of the cleavage site.
The user can choose whether to run SignalP-NN, SignalP-HMM, or both.
- Retraining of the neural networks:
SignalP-NN in SignalP V2.0 is trained on a newer data set derived
from SWISS-PROT rel. 35 (instead of rel. 29 as in SignalP V1.1).
- Graphics integrated in the output:
SignalP V2.0 shows signal peptide and cleavage site scores for each
position as plots in GIF format on the output page. The plots provide
more information than the prediction summary, e.g. about possible
cleavage sites other than the strongest prediction.
- Signal peptide region assignment:
SignalP-HMM provides not only a prediction of the presence of a signal
peptide and the position of the cleavage site, but also an approximate
assignment of n-, h- and c-regions within the signal peptide. These are
shown in the graphical output as probabilities for each position being
in one of these three regions.
- Automatic truncation:
in SignalP V1.1, we recommended that you should submit only the
N-terminal part of each protein, not more than 50-70 amino acids.
SignalP V2.0 now offers to truncate your sequences automatically.
Main publication:
-
Prediction of signal peptides and signal anchors by a hidden
Markov model.
Henrik Nielsen and Anders Krogh.
Proceedings of the Sixth International Conference on Intelligent
Systems for Molecular Biology (ISMB 6),
AAAI Press, Menlo Park, California, pp. 122-130, 1998.
|
|
1.1
|
The original server: the method based on artificial
neural networks.
Main publication:
-
Identification of prokaryotic and eukaryotic signal peptides
and prediction of their cleavage sites.
Henrik Nielsen, Jacob Engelbrecht, Søren Brunak
and Gunnar von Heijne.
Protein Engineering, 10:1-6, 1997.
|
Portable version
Would you prefer to run SignalP at your own site? SignalP 5.0 is available as a stand-alone software package, with the same functionality as this service. Ready-to-ship packages exist for Mac OS X (Darwin) and Linux platforms. There is a download page for academic users; other users are requested to contact DTU Health Technology Software Package Manager at
Software Downloads
- Version 6.0h
- Version 5.0b
- Version 4.1g
- Version 3.0
- Version 2.0