Neural networks
Artificial neural networks consist of a large number of independent computational units (so-called neurons) that are able to influence the computations of each other. A neuron has several inputs, and one output. The output from a neuron (a real number between 0 and 1) is calculated as a function of a weighted sum of the inputs. Several neurons can be connected (with the output of one neuron being the input of another neuron) thus forming a neural network. When a network is presented with an input (e.g. a string of real numbers that represent a sequence of amino acids) it will calculate an output that can be interpreted as a classification of the input (e.g. ``is the input sequence a signal peptide or not?''). It is possible to ``teach'' a neural network how to make a classification by presenting it with a set of known inputs (the training set) several times, and simultaneously modifying the weights associated with each input in such a way that the difference between the desired output and the actual output is minimized.
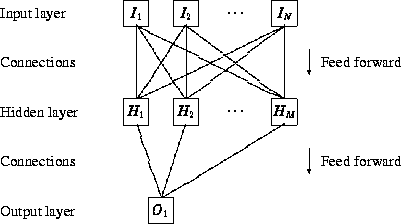
Simple neural networks of the kind used here (feed-forward networks) are closely related to the weight matrix method (von Heijne 1986), the two main differences being (1) that the weights in neural networks are found by training rather that statistical analysis, and (2) that neural networks are able to solve non-linear classification problems by introducing a layer of ``hidden neurons'' between input and output.
In this study, the output layer consisted of only one neuron which classified the sequence windows in two ways: Cleavage sites vs. other sequence positions, and signal peptide vs. non-signal-peptide. In the latter case, negative examples included both the first 70 positions of non-secretory proteins, and the first 30 positions of the mature part of secretory proteins.
For each of the five data sets and two classification problems, we tested several networks
with different numbers of input positions and hidden units, and selected the smallest
network that reached the optimal performance. While cleavage site networks worked best with
asymmetric windows (i.e. windows including more positions upstream than downstream of the
cleavage site), signal peptide networks work best with symmetric windows.
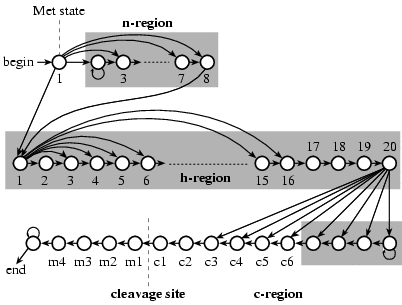
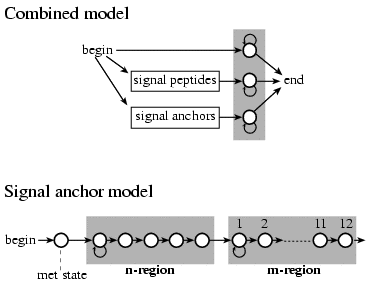
As a second machine learning algorithm method, a hidden Markov model (HMM) was developed in SignalP version 2.
The HMM also includes prediction of signal anchors in addition to the prediction of signal peptides. The figure
below show the architechture of the HMM.
Hidden Markov Models