Extraction
In SWISS-PROT sequences with 'SIGNAL' in the FT line were extracted. Only sequence entries with experimental evidence were used and signal peptide entries marked 'HYPOTHETICAL', 'POTENTIAL', 'PROBABLE' were removed. Furthermore were entires with more than one cleavage site removed. From secretory proteins, the sequence of the signal peptide and the first 30 amino acids of the mature protein were included in the data set. From cytoplasmic and (for the eukaryotes) nuclear proteins, the first 70 amino acids of each sequence were used. Additionally, a set of eukaryotic signal anchor sequences, i.e. N-terminal parts of type II membrane proteins, were extracted.
Redundancy in the data sets was avoided by excluding pairs of sequences which were functionally homologous, i.e. where the cleavage site of one signal peptide could be located by simply aligning it to the other. The numbers of sequences remaining in the final data sets (version 3) are shown below:
| Source | Number of sequences | |||
|---|---|---|---|---|
| Signal peptides | Cytoplasmic proteins | Signal anchors | Nuclear proteins | |
| Eukaryotes | 1192 | 459 | 67 | 990 |
| Gram- | 358 | 334 | - | - |
| Gram+ | 153 | 151 | - | - |
The redundancy reduction is described in detail in
"Defining a similarity threshold for a functional protein sequence
pattern: The signal peptide cleavage site",
Henrik Nielsen, Jacob Engelbrecht, Gunnar von Heijne, and
Søren Brunak,
Proteins, 24, 165-177, 1996.
The abstract can be found in the Abstract section
Spurious residues at position -1 was also removed in the training
set. Only amino acids A,L,G,xxx were alowed at this position.
To visualize the sequence information content in signal peptides,
we have generated sequence logos for the eukaryotic, Gram
negative and Gram positive training set.
The total height of the stack of letters at each position shows
the amount of sequence conservation at the position, while the relative
height of each letter shows the relative abundance of the corresponding
amino acid.
Sequence logos
of signal peptides, aligned after their cleavage sites.
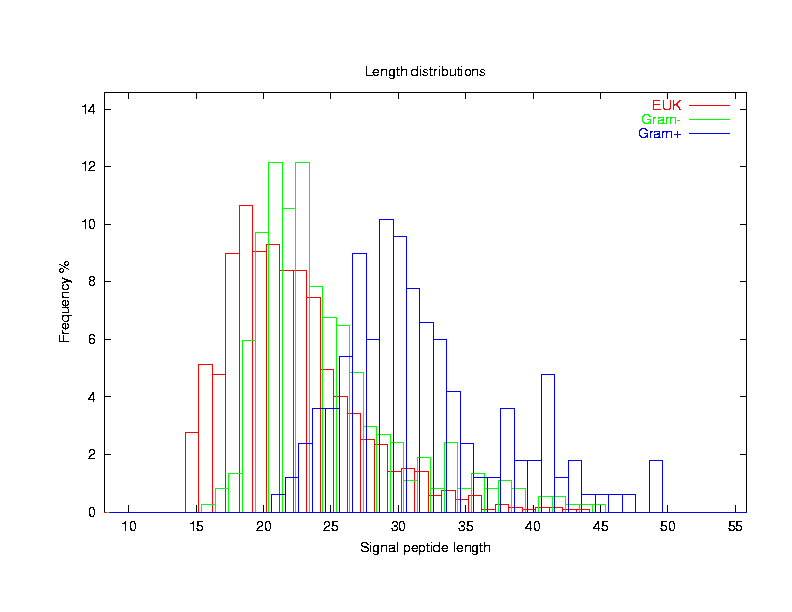
Distribution of lengths of the eukaryotic and prokaryotic signal peptides. The
average length is 22.6 amino acids for eukaryotes, 25.1 for Gram-negative
bacteria, and 32.0 for Gram-positive bacteria.
The common structure of signal peptides from various proteins is
commonly described as a positively charged n-region, followed by
a hydrophobic h-region and a neutral but polar c-region.
The (-3,-1)-rule states that the residues at positions -3 and -1
(relative to the cleavage site) must be small and neutral for cleavage
to occur correctly.
We have analyzed the characteristics of the new signal peptide data set and
displayed them in the form of sequence logos as previously shown. The
differences between signal peptides from different organisms are apparent from
the sequence logos:
Data set cleanup
Isoelectric point
We were able to identify wrongly annotated signal peptides from
Swiss-Prot simply by calculating the isoeletric point of the
signal peptide and corresponding mature protein.
Using this approach we were even able to identify wrongly
annotated start codons. Indepth information on this issue can be
found in "Improved prediction of signal peptides - SignalP 3.0 ", Bendtsen et al., 2004. The abstract can
be found in the Abstract section.
Propeptide
In Swiss-Prot, annotated signal peptide cleavage sites are entries found do in some
cases contain propeptides. These sequence entries were
reannotated acording to the output of SignalP version 2.
Propeptides have basic residues (R/K) at their cleavage sites.
Wrong start codons
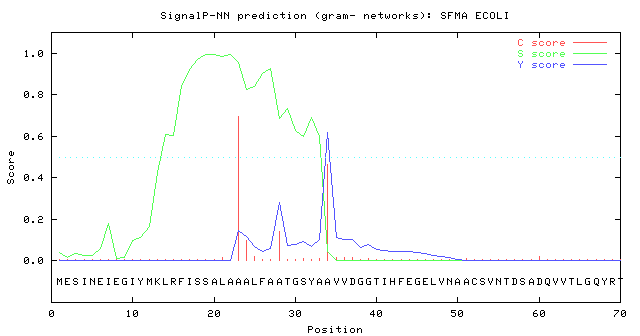
In specific cases, SignalP can be used as a "validation" tool for correct startcodon annotation. In
Swiss-Prot entry SFMA_ECOLI (Swiss-Prot rel. 40) the annotated Signal peptide cleavage site was
positioned 22 and a predicted cleavage site at position at 34. As the S-score (indicating signal
peptide-ness) is very low until position 11 we believed that the methionine at position 12 is the true
start of the protein. In a later release of Swiss-Prot this was indeed verified.
Sequence logos
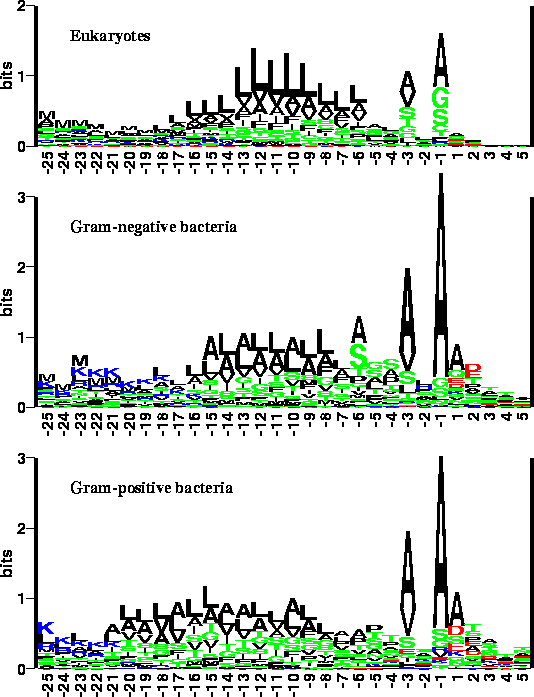
Blue:
Positively charged residues
Red:
Negatively charged residues
Green:
Neutral polar residues
Black:
Hydrophobic residues
Length distributions of signal peptides
Characteristics of signal peptides