Seq2Logo is a web-based sequence logo generation method for construction and
visualization of amino acid binding motifs and sequence profiles including
sequence weighting, pseudo counts and two-sided representation of amino acid
enrichment and depletion.
Note that Seq2Logo as default includes a pseudo count correction for lowcounts.
This means that the amino acid frequencies displayed in the sequence logos
are corrected for low number of observations using a Blosum amino acid
similarity matrix. To turn this feature off, the Weight on prior must be set to
zero.
Submission
CITATIONS
For publication of results, please cite:
Seq2Logo: a method for construction and visualization of amino acid binding
motifs and sequence profiles including sequence weighting, pseudo counts and
two-sided representation of amino acid enrichment and depletion.
Martin Christen Frolund Thomsen; Morten Nielsen, Nucleic Acids Research 2012; 40
(W1): W281-W287. Abstract Full text
NOTE
For big submissions please keep in mind that the computation time scales exponentially.
Eg.
A job of 10000 sequences with a sequence lenght of 38 takes about 20 seconds.
A job of 20000 sequences with a sequence lenght of 38 takes about 80 seconds.
A job of 40000 sequences with a sequence lenght of 38 takes about 6 minuts.
A job of 80000 sequences with a sequence lenght of 38 takes about 24 minuts.
etc.
if you submit too large alignments, the job might not finish within the server's time limit of 2 hours. To get results from these large submission you can download a local version of Seq2Logo and run it on your own machine.
Usage Instructions
The user interface of Seq2Logo is split in 3 parts; submission, graphical layout and advanced settings.
Submission
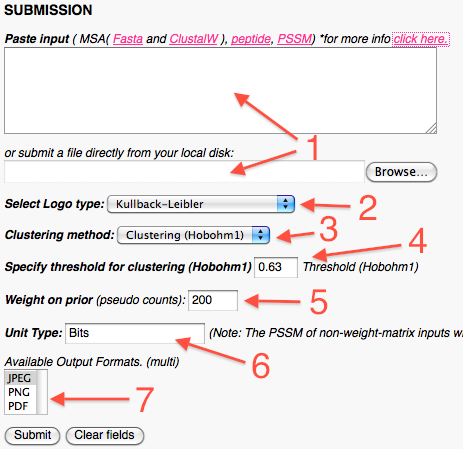
In the submission part the user can:
Upload their alignment file, either by copy/paste or by choosing a local file.
Specify the logo type, either Shannon, Kullback-Leibler, Weighted Kullback-Leibler, Probability Weighted Kullback-Leibler or PSSM-Logo.
Choose which kind of sequence weighting should be used to reduce sequence redundancy.
If the Hobohm algorithm is chosen, the user can also specify the similarity threshold for two sequences to be deemed (1 is equal 100% identity, default is 63%).
Assign the weight on prior value that should be used to adjust for a small alignment file (Recomended for dataset with less than 50 sequences).
Type the unit of the Y-axis. (It is important to note that MSA and rawpeptide input data will always be calculated as bit content *)
Choose additional output formats for the logo file.
* Shaner et al. gives a good description of the information content (the bit content) in their paper 'Sequence Logos: A Powerful, Yet Simple, Tool ', which can be accessed Here.
NOTE: As stated above the paste field Seq2Logo supports following alignments formats: Peptides, Fasta, Clustal,
weight matrices and frequency tables.
FORMAT DESCRIPTIONS:
The Peptide format is a file where each line is a new peptide sequence, only the amino acid and gap symbols are
accepted.
The Fasta format is a file where '>' describes the header line, and all following lines composes the sequence belonging
to the header. Only the amino acid and gap symbols are accepted in the sequence.
The CLUSTAL format is a file where the data is separated in two or three columns, first column containing the sequence
name, second column containing the sequence, and the optional third column containing the position number of the last amino
acid.
The PSSM format is a file where the data is stored in a weight matrix. There are a few different formats accepted by Seq2Logo:
General for all PSSM is the optional header line (starting with: 'Last position-specific scoring matrix...'),
and the required amino acid header line (this can now contain other character if the PSSM-logo is chosen).
In regards to the weights in the PSSM, only numbers (integers, floats and scientific), are allowed.
Simple Weight Matrix: This is the simplest of the weight matrices,
with only the weights provided. (Note: These weights cannot be integers!)
Weight Matrix w/ position: This is the same as the simple matrix,
but with the first column specifying the position (Note: Integers allowed!)
Weight Matrix w/ position and consensus sequence: This is the same
as the position matrix, but with an aditional column specifying the consensussequence (Note: This extra column is not used
by Seq2Logo, but only allowed for the convenience.)
Special Weight Matrix: This is a scrapped version of the simple matrix, and it
allows the user to specify other than amino acids eg. gaps. (Note: This matrix can only be used with the PSSM-logo option,
and there is a limitation of minimum 3 characters and maximum 20 characters!)
The Frequency format is identical to a PSSM-matrix, but where
weights/frequencies sums up to 1.00 per position (up to 2% inaccuracy allowed), and where of cause no weight/frequency is negative.
Graphical Layout
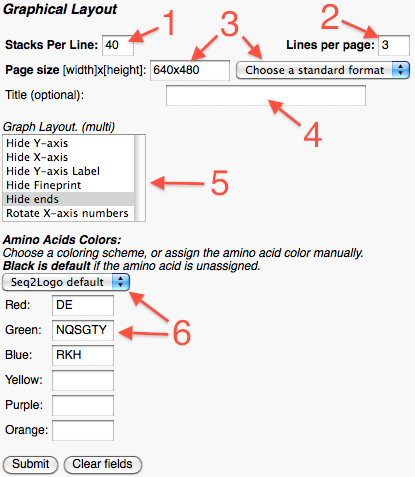
In the grafical layout part the user can:
Assign the number of stacks per line.
Assign the number of lines per page.
Set the resolution of the image. For convenience a dropdown menu has been provided with som standard formats to choose from.
Assign a logo title. (This is optional.)
Specify the layout of the graph. **
Choose a coloring scheme from the list, or assign the colors of the individual amino acids manually.
*
Feel free to send an email request if you want additional formats added.
**
This field allows you to really customize your logo.
Advanced Settings
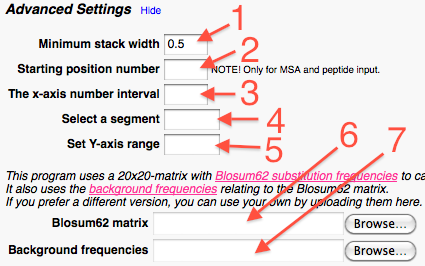
In the advanced settings part the user can:
Set the minimum width for stacks with gaps. * **
Set the position number of the first amino acid in the alignment.**
Set the frequency of which the position numbers are shown on the X-axis. ***
Set a segment range, if only a part of the full alignment is wanted. ****
Set the Y-axis range, This option allows the user to manually set the Y-axis maximum and minimum value, which makes it easier to compare several logos with eachother. *****
Upload separate substitution frequency matrix.
Upload separate Background frequency file (distribution of amino acids).
*
If set to 1 there is no width adjustment of the stacks to show positions where gaps occur.
**
This feature is meant for MSA and rawpeptide formats only.
***
If the value is set to 1 all positions numbers are shown. If the value is left out or 0 the interval is determined automatically.
****
Use the following format is "start-end", eg. 5-56
*****
Use the following format: "Ymin:Ymax", eg. -4.32:4.32
Implementation of easy access to Seq2Logo from other servers
Learn how to make an easy transfer of alignment files from your program or webpage to Seq2Logo Click here.
Output Format
DESCRIPTION
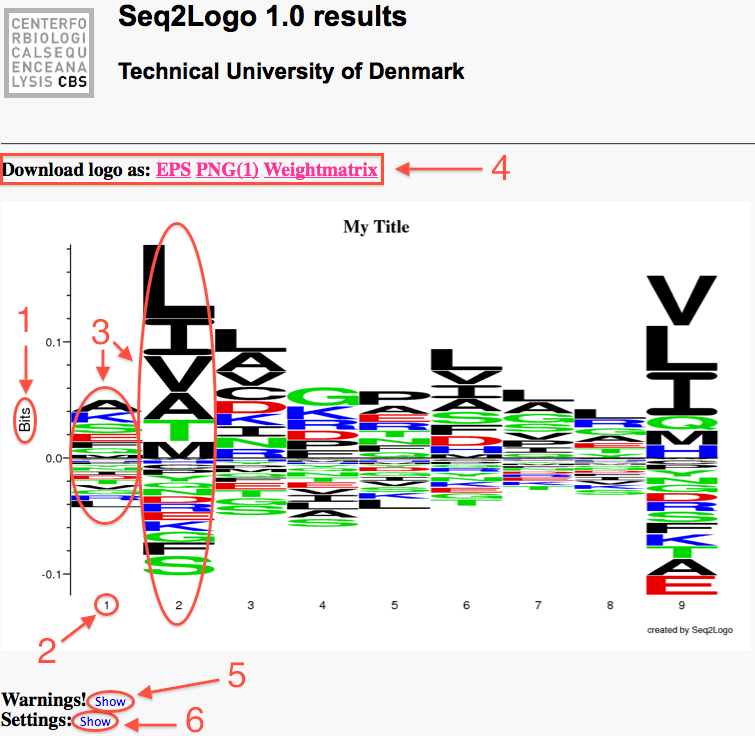
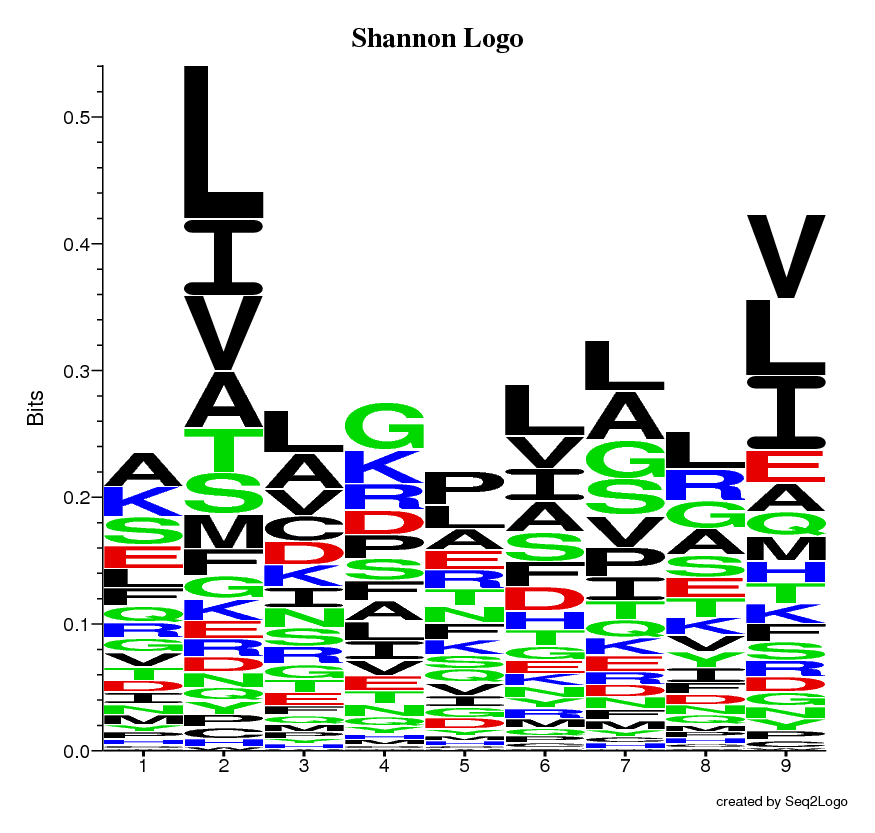
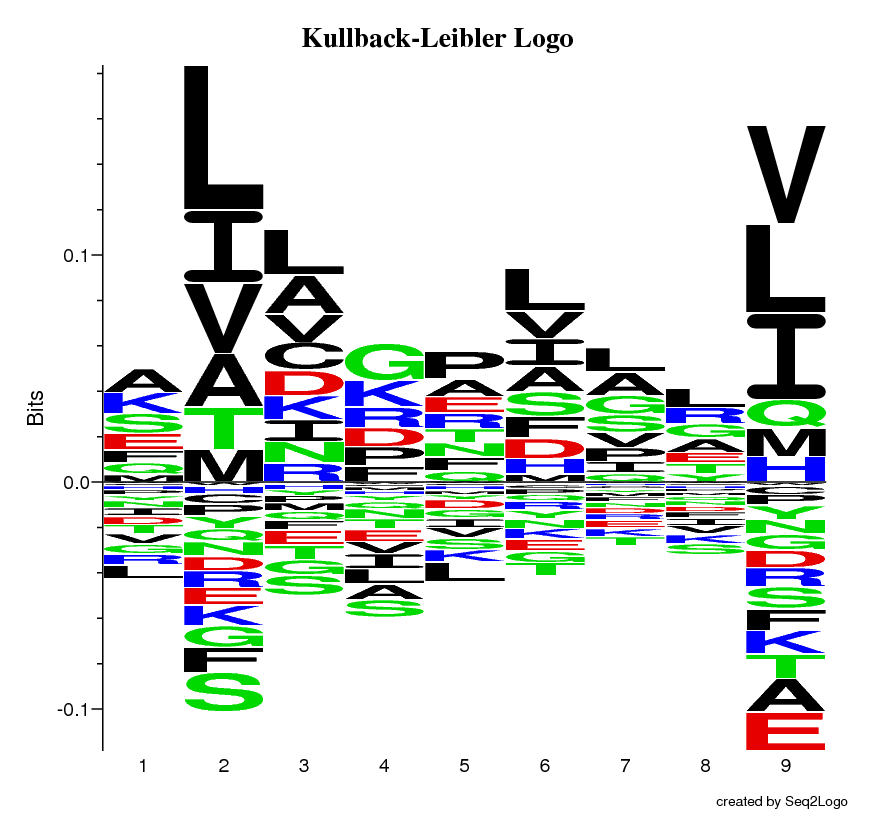
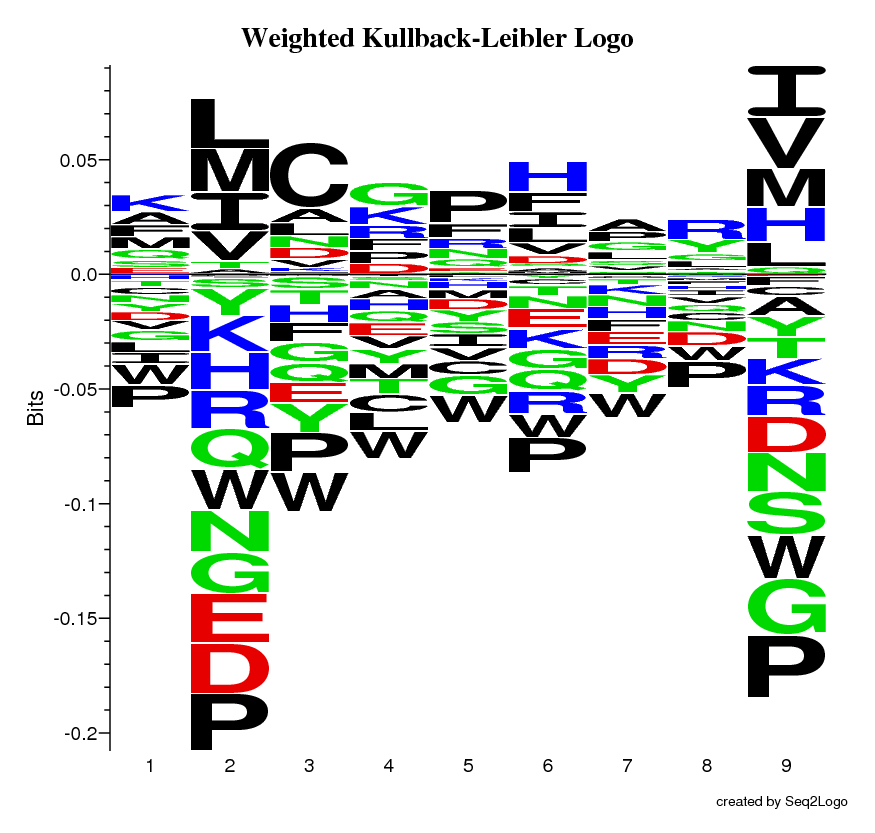
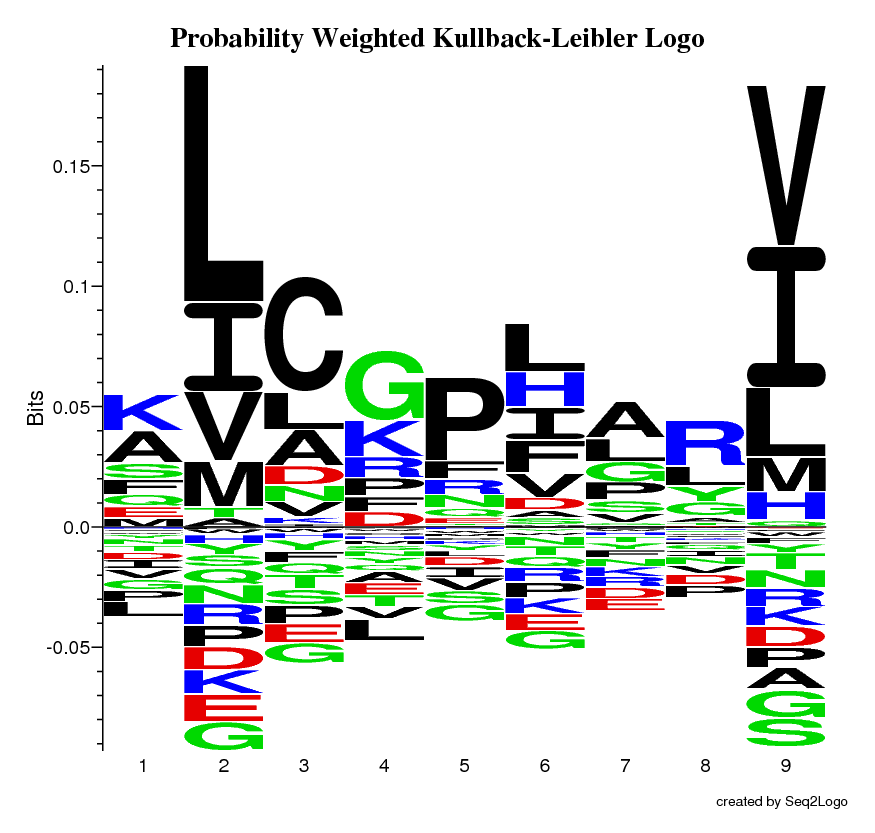
Once the Seq2Logo server has finished running the job you submitted it will show an image file containing the logo. This logo describes the information content of the alignment file you submitted.
The Y-axis describes the amount of information in bits*.
The X-axis shows the position in the alignment.
At each position there is a stack of symbols representing the amino acid. Large symbols represent frequently observed amino acids, big stacks represents conserved positions and small stacks represents variable positions.
The chosen formats plus the raw eps and the weight matrix is downloadable through the links in the top left corner.
By clicking the "show" link next to the "Warning!" sign, a list of the warnings will be shown. This will tell the user if any problem occurred, which might compromise the quality of the logo.
By clicking the "show" link next to the "Settings:" sign, a list of the user specified settings, which was used in the creation of the logo, will be shown.
* You can rename the Y-axis unit to what you prefer, but for all logos except of PSSM-logo the true unit is the bit content.
EXAMPLE OUTPUT
There are multiple logo types which influence the visual output. Click to show these different outputs here:
There are also a few methods which influence the visual output. These different outputs are shown here.
An alignment with gaps is handled by ignoring the gaps when calculating frequencies, and by shrinking the width of the stack according to the gap percentage.
The Shannon Logo
The Kullback-Leibler Logo, this logo type is the default of Seq2Logo. It gives a clear visual image of the conserved and variable regions, while also depicting depleted amino acids.
The Weighted Kullback-Leibler Logo, focusses on providing more information about the amino acid enrichment and depletion, instead of the probability, by altering the height of the individual amino acid to correspond to their log-odds score.
The Probability Weighted Kullback-Leibler Logo, this logo is a mixture of regular Kullback-Leibler and Weighted Kullback-Leibler, where the height of the amino acids corresponds to their probability times their log-odds score, which is also the information contribution of the amino acid. This logo really emphasizes the important parts.
The PSSM Logo, this logo type depicts the scoring matrix directly as it is, with no regards to the information centent. This logo has the advantage of clearly visualizing amino acid depletion at unconserved regions, and can be usefull to locate specific depletions such as the N-linked glycosylation sites with motif N-X-S/T, where X can be any amino acid but proline.
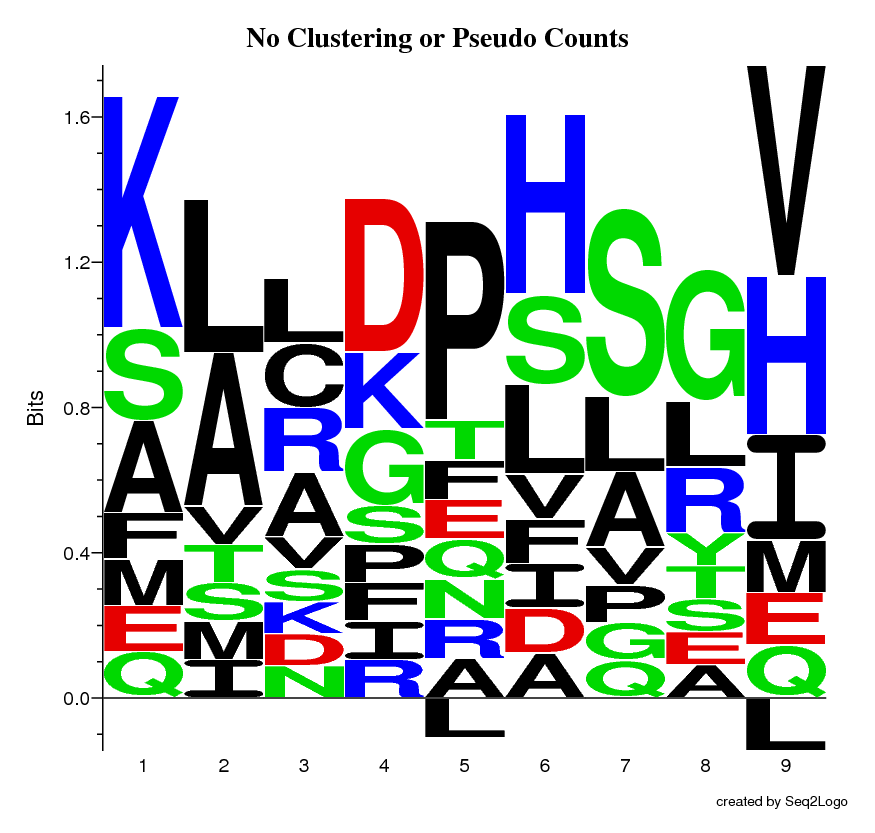
The Raw Logo, this shows a clean Kullback-Leibler logo without use of neither sequence weighting nor pseudo counts.
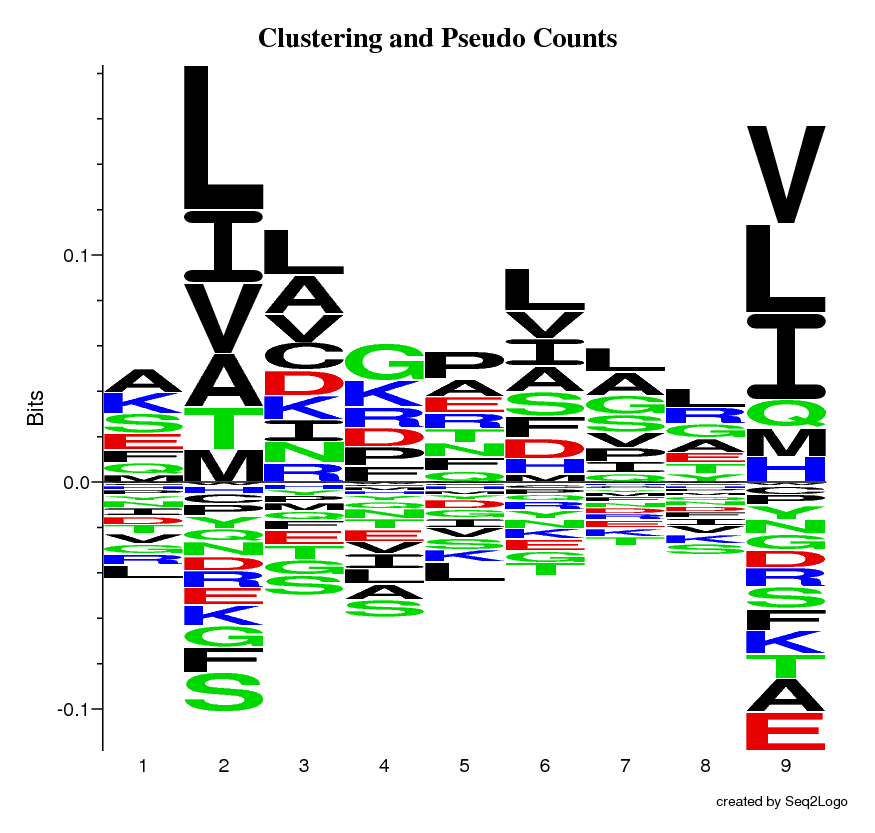
This logo is the same as the raw logo, but the redundancy in dataset has now been reduced by the use of the Hobohm 1 algorithm (sequence weighting method).
This logo is the same as the sequence weighted logo, but a pseudo counts method has been applied to correct for the lack of occurrences, due to a small dataset.
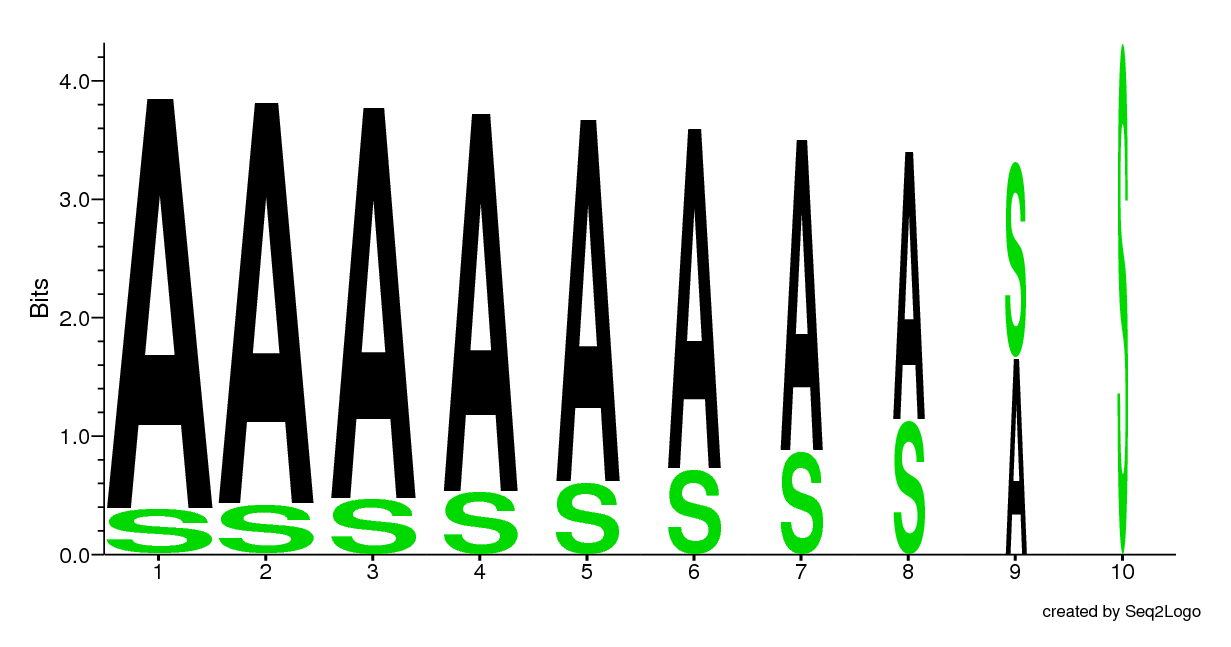
This logo shows the effect (shrinks the width of the stack) of gaps starting with 0% gaps and ending up with 90% gaps. In advanced options the user has the ability to set a minimum stack width. If this is set to 1 it will disable the shrinking effect. The default value is 0.5 (50%).
Article Abstract
Seq2Logo: A method for construction and visualization of amino acid
binding motifs and sequence profiles including sequence weighting, pseudo
counts and two-sided representation of amino acid enrichment and depletion.
and ,
DTU Health Tech, Technical
University of Denmark, DK-2800 Kgs Lyngby, Denmark.
Seq2Logo is a web-based sequence logo generator. Sequence logos are a graphical
representation of the information content stored in a multiple sequence
alignment (MSA) and provide a compact and highly intuitive representation of the
position-specific amino acid composition of binding motifs, active sites, etc.
in biological sequences. Accurate generation of sequence logos is often
compromised by sequence redundancy and low number of observations. Moreover,
most methods available for sequence logo generation focus on displaying the
position-specific enrichment of amino acids, discarding the equally valuable
information related to amino acid depletion. Seq2logo aims at resolving these
issues allowing the user to include sequence weighting to correct for data
redundancy, pseudo counts to correct for low number of observations and
different logotype representations each capturing different aspects related to
amino acid enrichment and depletion. Besides allowing input in the format of
peptides and MSA, Seq2Logo accepts input as Blast sequence profiles, providing
easy access for non-expert end-users to characterize and identify functionally
conserved/variable amino acids in any given protein of interest. The output from
the server is a sequence logo and a PSSM. Seq2Logo is available at
http://www.cbs.dtu.dk/biotools/Seq2Logo.
If you need help regarding technical issues (e.g. errors or missing results) contact Technical Support. Please include the name of the service and version (e.g. NetPhos-4.0) and the options you have selected. If the error occurs after the job has started running, please include the JOB ID (the long code that you see while the job is running).
If you have scientific questions (e.g. how the method works or how to interpret results), contact Correspondence.