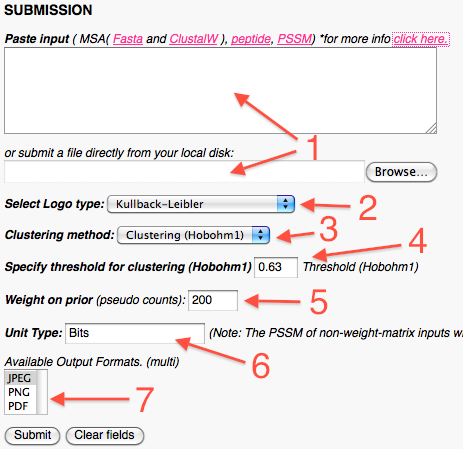
In the submission part the user can:
NOTE: As stated above the paste field Seq2Logo supports following alignments formats: Peptides, Fasta, Clustal, weight matrices and frequency tables.
FORMAT DESCRIPTIONS:The Peptide format is a file where each line is a new peptide sequence, only the amino acid and gap symbols are accepted.
The Fasta format is a file where '>' describes the header line, and all following lines composes the sequence belonging to the header. Only the amino acid and gap symbols are accepted in the sequence.
The CLUSTAL format is a file where the data is separated in two or three columns, first column containing the sequence name, second column containing the sequence, and the optional third column containing the position number of the last amino acid.
The PSSM format is a file where the data is stored in a weight matrix. There are a few different formats accepted by Seq2Logo:
General for all PSSM is the optional header line (starting with: 'Last position-specific scoring matrix...'),
and the required amino acid header line (this can now contain other character if the PSSM-logo is chosen).
In regards to the weights in the PSSM, only numbers (integers, floats and scientific), are allowed.
The Blast Matrix: Special format.
Simple Weight Matrix: This is the simplest of the weight matrices,
with only the weights provided. (Note: These weights cannot be integers!)
Weight Matrix w/ position: This is the same as the simple matrix,
but with the first column specifying the position (Note: Integers allowed!)
Weight Matrix w/ position and consensus sequence: This is the same
as the position matrix, but with an aditional column specifying the consensussequence (Note: This extra column is not used
by Seq2Logo, but only allowed for the convenience.)
Special Weight Matrix: This is a scrapped version of the simple matrix, and it
allows the user to specify other than amino acids eg. gaps. (Note: This matrix can only be used with the PSSM-logo option,
and there is a limitation of minimum 3 characters and maximum 20 characters!)
The Frequency format is identical to a PSSM-matrix, but where weights/frequencies sums up to 1.00 per position (up to 2% inaccuracy allowed), and where of cause no weight/frequency is negative.
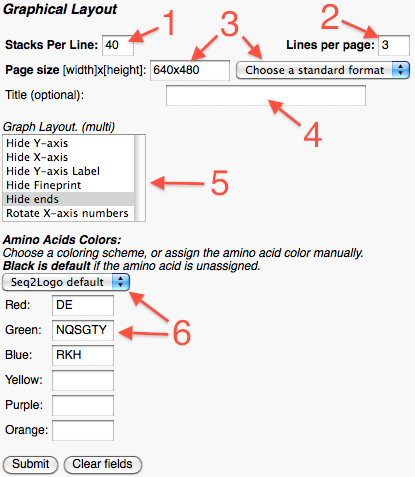
In the grafical layout part the user can:
| * | Feel free to send an email request if you want additional formats added. |
| ** | This field allows you to really customize your logo. |
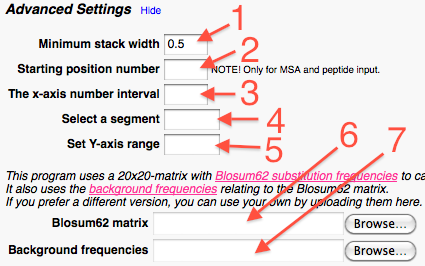
In the advanced settings part the user can:
| * | If set to 1 there is no width adjustment of the stacks to show positions where gaps occur. |
| ** | This feature is meant for MSA and rawpeptide formats only. |
| *** | If the value is set to 1 all positions numbers are shown. If the value is left out or 0 the interval is determined automatically. |
| **** | Use the following format is "start-end", eg. 5-56 |
| ***** | Use the following format: "Ymin:Ymax", eg. -4.32:4.32 |