Instructions & Guidelines
PopCover can be used to select MHC epitopes with optimal coverage of pathogen strains and a given population's HLA alleles. The method uses a scoring-based approach, where the highest scoring peptide is selected in each round.
What's new in version 2.0?
- PopCover-2.0 accepts both MHC class I and class II binders, which are combined into a single reduced dataset by the use of the Hobohm 1 algorithm.
- Optionally, HLA binders can be mapped onto equal-length peptides extracted from protein sequences that the binders originate from, thus increasing the possible number of alleles covered by each peptide.
- An accurate calculation of HLA loci coverage is included, both within and across loci.
- Visualization tables of the selected peptides can be generated, which provide a quick overview of the allele and genotype coverage.
1. Specify INPUT files
PopCover-2.0 needs at least one input file with MHC binders. Both MHC class I and class II binders can be uploaded.
An allele frequency list is required, which contains a list of HLA alleles that are present in the input epitope data, along with their frequencies. Allele frequencies can be downloaded from the
Allele Frequency Net Database.
Finally, the protein sequences from which the HLA binders originate from can be uploaded, either in protein FASTA format or simply with one protein sequence per line. If these sequences are supplied, the sequences will be chopped up into all unique peptides of a set length
n, and the input HLA binders will then be mapped onto these
n-mers. Any
n-mer which contains a given HLA binder as a substring will inherit the HLA binder's allele, genotype and binding core information. This will result in a list of equal-length peptides all containing some number of HLA binders each, and this list will then be reduced by the Hobohm 1 method. Afterwards, the peptides with widest coverage will be selected. Note that running in this mode can take longer due to the mapping onto the
n-mers.
Note that submitting the protein sequences is completely optional.
If no protein sequences are supplied, the initial peptide list will simply be the set of unique input HLA binders, which can all vary in length. As before, this initial list will then be reduced using the Hobohm 1 method, and the peptides with widest coverage will then be selected.
You can choose to either paste your data lines directly into the textboxes, or upload the files from your local disk.
2. Set OPTIONS to customize your analysis
The numerical input options are:
- Number of epitopes to select - an integer value.
- Peptide length to extract from protein sequences - an integer value. Input protein sequences will be chopped up into peptides of the length specified in this option. Make sure that this number is larger than or equal to the largest length of your HLA binders.
- Number of epitope sets to select - an integer value. The epitope selection scheme will be repeated this number of times, which will yield different sets of peptide selections.
- Offset for denominator in score function (β) - a tunable constant bigger than 0. Large β values put more emphasis on the individual peptide's allelic coverage and less on what has already been covered by previous selections.
- Minimum genomic coverage - a float value between 0 and 1. This is a threshold for the fraction of pathogen strains peptides must cover in order to be considered.
By default, the standard PopCover selection scheme is used. Here, the first peptide will be selected based on the initial score given by
Sini = (CI + CII)*G
where
CI is the peptide's coverage across MHC class I alleles,
CII is the peptide's coverage across MHC class II alleles, and
G is the peptide's number of covered genotypes. The remaining peptides are selected based on the PopCover scoring function which optimizes allelic and genotypic coverage with each selection.
Alternatively, you can use one of these selection methods:
- Rank peptides based on their initial scores and select the top n peptides - Peptides will be selected solely based on their inital scores Sini.
- Select peptides at random - use completely random selection instead of the PopCover scoring scheme. Included for benchmarking purposes.
The binary options are:
- MHC class I/II - no binding core column - select these options if either of your MHC class I and class II binder datasets do not contain a binding core column. This is to prevent the genotype column from being interpreted as the binding core column. If no binding core column is supplied, the peptide itself will be treated as a binding core.
- Use phenotypic frequencies for calculation - the phenotypic frequency q is used instead of the allelic frequency f. The relevant formula is q = 2·f −f2
- Subtract min. genomic coverage in score function denominator - if checked, the minimum genomic coverage must not be equal to the denominator offset value β, in order to avoid division by zero.
- Skip Hobohm 1 dataset reduction - if checked, the dataset of peptides will not be reduced by the Hobohm 1 algorithm. It is recommended to leave this option unchecked, to avoid selection of redundant peptides.
- Require nested class I/II binder in selected peptides - if either of the two options are checked, the selected peptides will be required to contain a nested class I and/or class II binder.
- Use IEDB's method for calculating population coverage - if checked, the population coverage will be calculated with the method used in IEDB's Population Coverage tool. This may yield slightly higher coverage estimates than the standard method used in PopCover-2.0.
3. SUBMIT the job
Click on the "Submit" button. The status of your job (either 'queued' or 'running') will be displayed and constantly updated until it terminates and the server output appears in the browser window.
At any time during the wait you may enter your e-mail address and simply leave the window. Your job will continue; you will be notified by e-mail when it has terminated. The e-mail message will contain the URL under which the results are stored; they will remain on the server for 24 hours for you to collect them.
How input data is handled
If protein sequences are supplied, equal-length peptides are extracted from them, and the input HLA binders are then mapped onto these n-mer peptides. If a binder peptide is contained as a substring in an n-mer peptide, the n-mer peptide 'inherits' the binder's covered alleles, genotypes and their combinations. The list of unique n-mer peptides with nested HLA binders is then reduced using the Hobohm 1 algorithm.
If no protein sequences are supplied, the HLA binders are read in as normal, and they will form the initial peptide list to be reduced with Hobohm 1. Note that this list can have peptides in varying lengths.
How does the Hobohm 1 dataset reduction work?
Hobohm 1 takes as input a list of peptides sorted in descending order of importance, and iteratively compares the peptide at the top of the list to a ’unique’ peptide list. If a peptide is found to be similar to any peptide in the unique list, it is discarded, otherwise it is added to the unique list.
If the two peptides being compared have different lengths (i.e. no protein sequences were supplied for extracting equal-length
n-mers), it is checked if one is a substring of the other. If this is the case, the longer peptide inherits the shorter peptide's allele, genotype and binding core information, and the shorter peptide is then discarded. However, if no substring is found or the peptides have equal length (meaning no substring can be found), another criterion is checked. A given peptide is deemed redundant if its
coverage set is a subset of the unique peptide’s coverage set. Here, a coverage set is a set of unique binding core, allele, genotype combinations. An example could be
{ (core
1, hla
1, genotype
1) , (core
1, hla
2, genotype
1) }
The rationale here is that if peptide A’s coverage set is a superset of peptide B’s coverage set,
peptide A can perform the same function as peptide B, and peptide B is thus redundant.
The input peptide list to Hobohm 1 is sorted in descending order first by length, then by the sizes of the coverage sets, i.e. the number of allele, genotype, binding core combinations. In this way, the longest peptides and peptides with the widest coverage will be a the top of the list, and will be more likely to be added to the unique list.
PopCover 2.0 - Output Format
The output of PopCover 2.0 contains the following elements:
Dataset overview
Number of unique peptides, alleles and genotypes found in the data.
Dataset reduction result
How many peptides the dataset contains before and after Hobohm 1 reduction.
List of alleles and genotypes in the dataset
The order of occurence of alleles and genotypes matches the counters included in the next section.
Sequential peptide selection
A vector of counters for each allele and genotype is displayed for each selected peptide. These counters shows the accumulated number of peptides which cover each allele and genotype. Additionally, the accumulated number of covered allele+genotype combinations is shown.
Peptide selection table
A table which contains the selected peptides and some metrics. The headers are the following:
- i_score - Initial score given to each peptide based on its individual coverage of MHC Class I and Class II. The formula is Sinitial = (hlacov_I + hlacov_II)*nhitg
- score - The score given to the peptide during selection. The first peptide's score will be equal to i_score, while the rest of the scores will be the ones given by the PopCover scoring function.
- hlacov_I - The MHC Class I coverage across loci for the individual peptide.
- hlacov_II - The MHC Class II coverage across loci for the individual peptide.
- nhita_I - Number of covered MHC Class I alleles.
- nhita_II - Number of covered MHC Class II alleles.
- nhitg - Number of covered genotypes.
Locus coverage
For each MHC class present in the data, a list of the individual loci within the class are listed, along with the calculated coverage both within and across the loci.
Number of covering peptides per allele and per genotype
The same numbers shown under the last peptide in the
Sequential peptide selection section.
Downloads
Overview of alleles and genotypes in the data, along with the selected peptides, will be available for download in .txt format.
Aditionally, a colored visualization of the
Sequential peptide selection will be available in .png and .xlsx formats, which illustrates how the coverage of the different alleles and genotypes changes with each peptide selection.
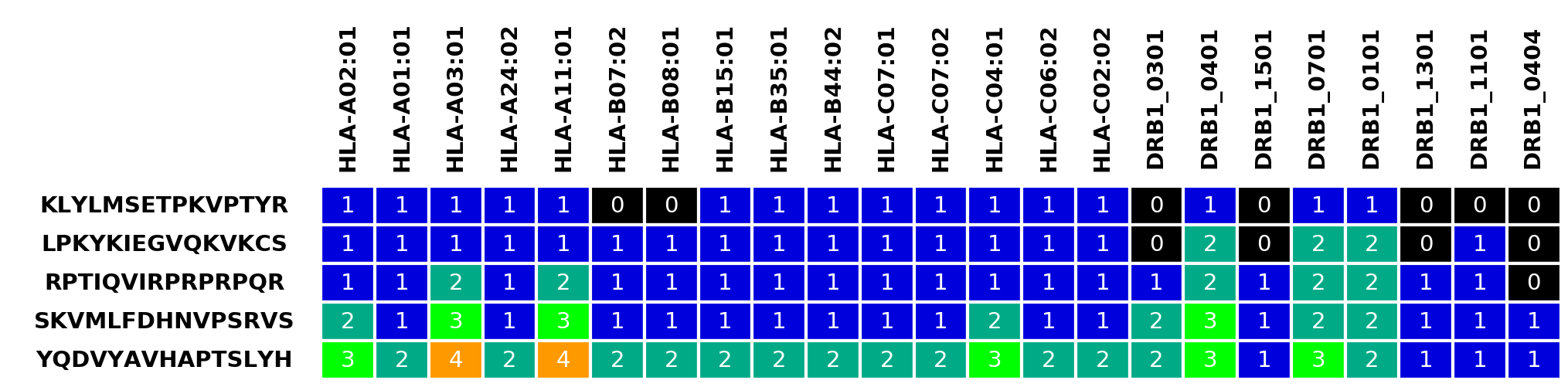
Example HLA table
Article Abstract
PopCover-2.0. Improved Selection of Peptide Sets With Optimal HLA and Pathogen Diversity Coverage
Jonas Birkelund Nilsson, Alba Grifoni, Alison Tarke, Alessandro Sette and Morten Nielsen
Frontiers in Immunology, 17 August 2021. Link to article
The use of minimal peptide sets offers an appealing alternative for design of vaccines and T cell diagnostics compared to conventional whole protein approaches. T cell immunogenicity towards peptides is contingent on binding to human leukocyte antigen (HLA) molecules of the given individual. HLA is highly polymorphic, and each variant typically presents a different repertoire of peptides. This polymorphism combined with pathogen diversity challenges the rational selection of peptide sets with broad immunogenic potential and population coverage. Here we propose PopCover-2.0, a simple yet highly effective method, for resolving this challenge. The method takes as input a set of (predicted) CD8 and/or CD4 T cell epitopes with associated HLA restriction and pathogen strain annotation together with information on HLA allele frequencies, and identifies peptide sets with optimal pathogen and HLA (class I and II) coverage. PopCover-2.0 was benchmarked on historic data in the context of HIV and SARS-CoV-2. Further, the immunogenicity of the selected SARS-CoV-2 peptides was confirmed by experimentally validating the peptide pools for T cell responses in a panel of SARS-CoV-2 infected individuals. In summary, PopCover-2.0 is an effective method for rational selection of peptide subsets with broad HLA and pathogen coverage. The tool is available at https://services.healthtech.dtu.dk/service.php?PopCover-2.0.
Software Download
Want to run PopCover-2.0 locally instead? An offline version of the code is available
here . You will need to install Python 3 (version 3.7+) including the
numpy library.