CITATIONS
For publication of results, please cite:
Machine learning reveals limited contribution of trans-only encoded variants to the HLA-DQ immunopeptidome
Jonas Birkelund Nilsson, Saghar Kaabinejadian, Hooman Yari, Bjoern Peters, Carolina Barra, Loren Gragert, William Hildebrand and Morten Nielsen
Communications Biology, 21 April 2023.
https://doi.org/10.1038/s42003-023-04749-7
PORTABLE VERSION
NetMHCIIpan 4.2 is available
as a stand-alone software package, with the same functionality as the service
above. Ready-to-ship packages exist for Linux and macOS. There is a
download page
for academic users; other users are requested to contact Health Tech Software Package
Manager at health-software@dtu.dk.
Instructions
INPUT DATA
In this section, the user must define the input for the prediction server following these steps:
1) Specify the desired type of input data (FASTA or PEPTIDE ) using the drop down menu.
2) Provide the input data by means of pasting the data into the blank field, uploading it using the "Choose File" button or by loading sample data using the "Load Data" button. All the input sequences must be in one-letter amino acid code. The alphabet is as follows (case sensitive):
A C D E F G H I K L M N P Q R S T V W Y and X (unknown)
Any other symbol will be converted to X before processing. At most 5000 sequences are allowed per submission; each sequence must be not more than 20,000 amino acids long and not less than 9 amino acids long.
3) If FASTA was selected as input type, the user must select the peptide length(s) the prediction server is going to work with. NetMHCIIpan-4.2 will "chop" the input FASTA sequence in overlapping peptides of the provided length and will predict binding against all of them. By default input proteins are digested into 15-mer peptides. Note that, if PEPTIDE was selected as input type, this step is unnecessary and thus the peptide length selector will directly not appear in the interface.
4) Context encoding informs the network of the proteolytic context the ligand. Context is automatically generated from the source protein if the user selects FASTA format. Briefly, context is made up of 12 amino acids: 3 amino acids upstream of the ligand, 3 first amino acids at the ligand N-terminus, 3 last amino acids at the ligand C-terminus and 3 amino acids downstream the ligand(in the source protein), all concatenated together. If the input type is PEPTIDE , the user must specify the ligand context(see PEPTIDECONT ).

MHC SELECTION
In this section, the user must define which MHC molecule(s) the input data is going to be predicted against:
1) Here the user can select from a list of MHC molecules by first selecting the group and clicking MHCs in the list. Here, MS-COVERED refers to molecules properly covered by the NetMHCIIpan-4.2 training data.
2) The user can also type the molecule names. Both the ALPHA and BETA chains must be typed (please consult List of MHC molecule names.) Note that molecules selected from step 1 populate this bar.
3) If the molecule of interest is not provided in the lists, the user can input ALPHA and BETA sequences in fasta format. With this option, rank score predictions are not available.
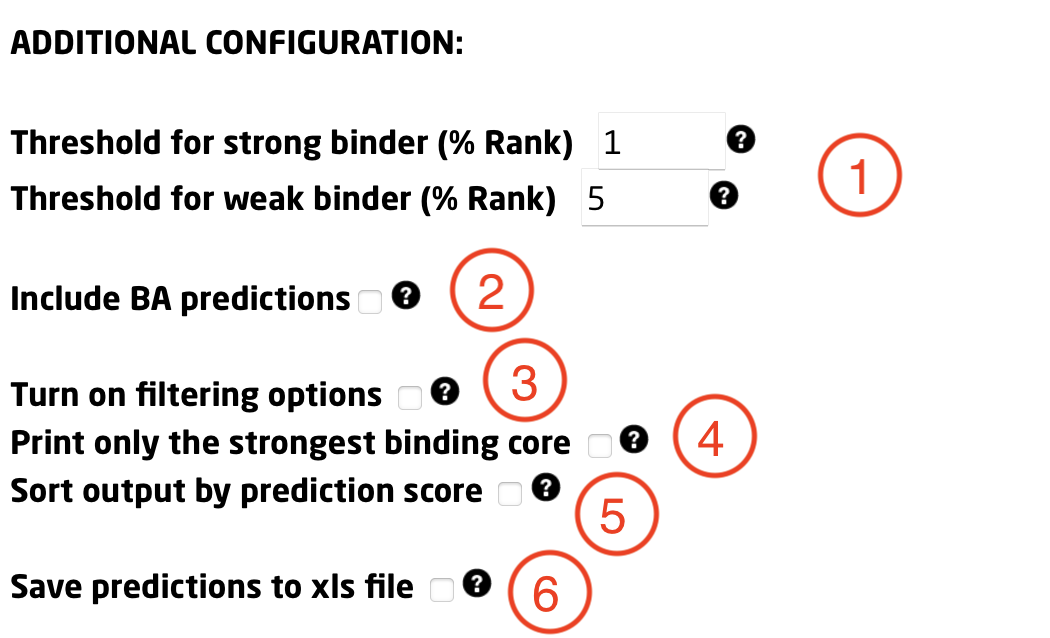
ADDITIONAL CONFIGURATION
In this section, the user may define additional parameters to further customize the run:
1) Specify thresholds for strong and weak binders. They are expressed in terms of %Rank, that is percentile of the predicted binding affinity compared to the distribution of affinities calculated on set of random natural peptides. The peptide will be identified as a strong binder if it is found among the top x% predicted peptides, where x% is the specified threshold for strong binders (by default 2%). The peptide will be identified as a weak binder if the % Rank is above the threshold of the strong binders but below the specified threshold for the weak binders (by default 10%).
2) Tick this option to include also Binding Affinity predictions together with Eluted Ligand likelihood.
3) Tick this option to output only peptides with a % Rank score below a specified threshold. Useful for large submissions.
4) Tick this box to output only the strongest binding core.
5) Tick this box to have the output sorted by descending prediction score.
6) Enable this option to export the prediction output to .XLS format (readable for most spreadsheet softwares, like Microsoft Excel).
SUBMISSION
After the user has finished the "INPUT DATA", "MHC SELECTION" and "ADDITIONAL CONFIGURATION" steps, the submission can now be done. To do so, the user can click on "Submit" to submit the job to the processing server, or click on "Clear fields" to clear the page and start over.
The status of your job (either 'queued' or 'running') will be displayed and constantly updated until it terminates and the server output appears in the browser window.
After the server has finished running the corresponding predictions, an
output page will be delivered to the user. A description of the output format can be found at outpur format
At any time during the wait you may enter your e-mail address and simply leave the window. Your job will continue; when it terminates you will be notified by e-mail with a URL to your results. They will be stored on the server for 24 hours.
Output format
EXAMPLE OUTPUT
For the following FASTA input example:
>P9WNK5
MAEMKTDAATLAQEAGNFERISGDLKTQIDQVESTAGSLQGQWRGAAGTAAQAAVVRFQEAANKQKQELDEISTNIRQAGVQYSRADEEQQQALSSQMGF
With parameters:
Peptide length: 15
Allele: DQA10301-DQB10302
Sort by prediction score: On
NetMHCIIpan-4.2 will return the following output (showing the top 10 predicted peptides):
# NetMHCIIpan version 4.2
# Input is in FASTA format
# Peptide length 15
# Prediction Mode: EL
# Threshold for Strong binding peptides (%Rank) 1%
# Threshold for Weak binding peptides (%Rank) 5%
# DQA10301-DQB10302 : Distance to training data 0.0000 (using nearest neighbor HLA-DQA10301-DQB10302)
# Allele: DQA10301-DQB10302
--------------------------------------------------------------------------------------------------------------------------------------------
Pos MHC Peptide Of Core Core_Rel Identity Score_EL %Rank_EL Exp_Bind BindLevel
--------------------------------------------------------------------------------------------------------------------------------------------
3 DQA10301-DQB10302 EMKTDAATLAQEAGN 4 DAATLAQEA 0.590 P9WNK5 0.467401 0.23 NA <=SB
78 DQA10301-DQB10302 QAGVQYSRADEEQQQ 3 VQYSRADEE 0.900 P9WNK5 0.418179 0.35 NA <=SB
4 DQA10301-DQB10302 MKTDAATLAQEAGNF 3 DAATLAQEA 0.770 P9WNK5 0.406498 0.38 NA <=SB
2 DQA10301-DQB10302 AEMKTDAATLAQEAG 5 DAATLAQEA 0.530 P9WNK5 0.381155 0.47 NA <=SB
77 DQA10301-DQB10302 RQAGVQYSRADEEQQ 4 VQYSRADEE 0.890 P9WNK5 0.348975 0.64 NA <=SB
5 DQA10301-DQB10302 KTDAATLAQEAGNFE 2 DAATLAQEA 0.790 P9WNK5 0.325364 0.79 NA <=SB
79 DQA10301-DQB10302 AGVQYSRADEEQQQA 2 VQYSRADEE 0.740 P9WNK5 0.295187 1.03 NA <=WB
1 DQA10301-DQB10302 MAEMKTDAATLAQEA 6 DAATLAQEA 0.420 P9WNK5 0.260940 1.41 NA <=WB
76 DQA10301-DQB10302 IRQAGVQYSRADEEQ 5 VQYSRADEE 0.960 P9WNK5 0.250306 1.54 NA <=WB
80 DQA10301-DQB10302 GVQYSRADEEQQQAL 1 VQYSRADEE 0.600 P9WNK5 0.160670 3.56 NA <=WB
DESCRIPTION
The prediction output for each molecule consists of the following columns:
Pos Residue number (starting from 0)
MHC MHC molecule name
Peptide Amino acid sequence
Of Starting position offset of the optimal binding core (starting from 0)
Core Binding core register
Core_Rel Reliability of the binding core, expressed as the fraction of networks in the ensemble selecting the optimal core
Identity Annotation of the input sequence, if specified
Score_EL Eluted ligand prediction score
%Rank_EL Percentile rank of eluted ligand prediction score
Exp_bind If the input was given in PEPTIDE format with an annotated affinity value (mainly for benchmarking purposes).
Score_BA Predicted binding affinity in log-scale (printed only if binding affinity predictions were selected)
Affinity(nM) Predicted binding affinity in nanomolar IC50 (printed only if binding affinity predictions were selected)
%Rank_BA % Rank of predicted affinity compared to a set of 100.000 random natural peptides. This measure is not affected by inherent bias of certain molecules towards higher or lower mean predicted affinities (printed only if binding affinity predictions were selected)
BindLevel (SB: strong binder, WB: weak binder). The peptide will be identified as a strong binder if the % Rank is below the specified threshold for the strong binders. The peptide will be identified as a weak binder if the % Rank is above the threshold of the strong binders but below the specified threshold for the weak binders.
Article abstracts
Machine learning reveals limited contribution of trans-only encoded variants to the HLA-DQ immunopeptidome
Jonas Birkelund Nilsson, Saghar Kaabinejadian, Hooman Yari, Bjoern Peters, Carolina Barra, Loren Gragert, William Hildebrand and Morten Nielsen
Communications Biology, 21 April 2023. https://doi.org/10.1038/s42003-023-04749-7
Human leukocyte antigen (HLA) class II antigen presentation is key for controlling and triggering T cell immune responses. HLA-DQ molecules, which are believed to play a major role in autoimmune diseases, are heterodimers that can be formed as both cis and trans variants depending on whether the α- and β-chains are encoded on the same (cis) or opposite (trans) chromosomes. So far, limited progress has been made for predicting HLA-DQ antigen presentation. In addition, the contribution of trans-only variants (i.e. variants not observed in the population as cis) in shaping the HLA-DQ immunopeptidome remains largely unresolved. Here, we seek to address these issues by integrating state-of-the-art immunoinformatics data mining models with large volumes of high-quality HLA-DQ specific mass spectrometry immunopeptidomics data. The analysis demonstrates highly improved predictive power and molecular coverage for models trained including these novel HLA-DQ data. More importantly, investigating the role of trans-only HLA-DQ variants reveals a limited to no contribution to the overall HLA-DQ immunopeptidome. In conclusion, this study furthers our understanding of HLA-DQ specificities and casts light on the relative role of cis versus trans-only HLA-DQ variants in the HLA class II antigen presentation space. The developed method, NetMHCIIpan-4.2, is available at https://services.healthtech.dtu.dk/services/NetMHCIIpan-4.2.
Supplementary material
Training data
NetMHCIIpan-4.2
Here, you will find the data set used for training of NetMHCIIpan-4.2.
NetMHCIIpan_train.tar.gz
Download the file and untar the content using
cat NetMHCIIpan_train.tar.gz | tar xvf -
This will create the directory called NetMHCIIpan_train. In this directory you will find 12 files. 10 files (c00?_ba, c00?_el) with partitions with binding affinity (ba) with eluted ligand data (el). The format for each file is (here shown for an el file)
AAAAAAAAAAAA 1 Saghar_9075 AGRAAAAAAAAA
AAAAAAAAAAAA 1 Saghar_9090 AGRAAAAAAAAA
AAAAAAAAAAAAA 1 Bergseng__9037_SWEIG AGRAAAAAAAAG
AAAAAAAAAAAAA 1 Bergseng__9064_AMALA AGRAAAAAAAAG
AAAAAAAAAAAAA 1 Bergseng__9089_BOB AGRAAAAAAAAG
AAAAAAAAAAAAA 1 Saghar_9052 AGRAAAAAAAAG
AAAAAAAAAAAAA 1 Saghar_9075 AGRAAAAAAAAG
AAAAAAAAAAAAA 1 Saghar_9090 AGRAAAAAAAAG
AAAAAAAAAAAAAA 1 Bergseng__9037_SWEIG AGRAAAAAAAGA
AAAAAAAAAAAAAA 1 Bergseng__9064_AMALA AGRAAAAAAAGA
where the different columns are peptide, target value, MHC_molecule/cell-line, and context. In cases where the 3rd columns is a cell-line ID, the MHC molecules expressed in the cell-line are listed in the allelelist.txt file.
The allelelist.txt file contains the information about alleles expressed in each MA cell line data set, and pseudosequence.2016.fix the MHC pseudo sequenes for each MHC molecule.
Version history
Please click on the version number to activate the corresponding server.
|
4.2
|
The current server (online since September 2022). New in this version:
- NetMHCIIpan-4.2 is trained on an extensive dataset of both eluted ligand (EL) and binding affinity (BA) data, including new novel EL data for 14 HLA-DQ molecules. Further, a 'distance to training data' metric is printed for each selected molecule in the same way as NetMHCpan-4.1, indicating how reliable the predictions are.
Main publication:
-
Machine learning reveals limited contribution of trans-only encoded variants to the HLA-DQ immunopeptidome
Jonas Birkelund Nilsson, Saghar Kaabinejadian, Hooman Yari, Bjoern Peters, Carolina Barra, Loren Gragert, William Hildebrand and Morten Nielsen
Communications Biology, 21 April 2023. https://doi.org/10.1038/s42003-023-04749-7
|
|
4.1
|
(online since Sept 2021). New in this version:
- The method is trained on a extented set of EL data compared to version 4.0, and novel and correct BA for HLA-DQA1*04:01-DQB1*04:02 are included. Further is DRB3, 4 and 5 allele information predicted from DRB1 based on linkage disequilibrium with DRB1 when absent from typing data.
Main publication:
-
Accurate MHC Motif Deconvolution of immunopeptidomics data reveals high relevant contribution of DRB3, 4 and 5 to the total DR Immunopeptidome
Saghar Kaabinejadian, Carolina Barra, Bruno Alvarez, Hooman Yari, William Hildebrand, Morten Nielsen
Frontiers in Immunology 26 January 2022. Sec. Antigen Presenting Cell Biology, DOI: 10.3389/fimmu.2022.835454
|
|
4.0
|
(online since April 2020). New in this version:
-
The two output neuron architechture introduced in NetMHCpan-4.0 permits the inclusion of EL data, and the new training algorithm NNAlign_MA extends training data to ligands of ambiguous allele assignments. The model also, optionally, encodes ligand context.
Main publication:
-
Improved prediction of MHC II antigen presentation through integration and motif deconvolution of mass spectrometry MHC eluted ligand data.
Reynisson B, Barra C, Kaabinejadian S, Hildebrand WH, Peters B, Nielsen M
J Proteome Res 2020 Apr 30. doi: 10.1021/acs.jproteome.9b00874.
PubMed: 32308001
|
|
3.2
|
(online since January 2018). New in this version:
- Method retrained on an extensive dataset of over 100,000 datapoints, covering 36 HLA-DR, 27 HLA-DQ, 9 HLA-DP, and 8 mouse MHC-II molecules.
Main publication:
-
Improved methods for predicting peptide binding affinity to MHC class II molecules.
Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, Sette A, Peters B, Nielsen M.
Immunology. 2018 Jan 6. doi: 10.1111/imm.12889.
PubMed: 29315598
|
|
3.1
|
(online since December 2014). New in this version:
- Improved binding core identification by realigning individual networks in the ensemble.
- Introduced a reliability measure on the predicted binding core (Core_Rel column).
- Graphical representation of the binding core register and of possible multiple cores.
Main publication:
-
Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification
Andreatta M, Karosiene E, Rasmussen M, Stryhn A, Buus S, and Nielsen M
Immunogenetics (2015)
PubMed: 26416257
|
|
3.0
|
(online since June 2013). New in this version:
- The user can make predictions for all DR, DP and DQ molecules with known protein
sequence. Likewise can the user upload full length MHC class II alpha and beta chain and have the server predict MHC restricted peptides from any given protein of interest
|
|
2.1
|
(online since 6 June 2011). New in this version:
- User can upload full length MHC class II beta chain and have the server predict MHC restricted peptides from any given protein of interest.
|
|
2.0
|
(online since 17 Nov 2010). New in this version:
- New concurent algorithm used to train the network.
|
|
1.1
|
(online since 15 April 2010). New in this version:
- %-rank measure include for each prediction value. The %-rank score give the rank of the
prediction score to a distribution of prediction scores from 200.000 natural random 15mer peptides.
|
|
1.0
|
Original version (online version until April 15 2010):
Main publication:
-
Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan.
Nielsen M, et al. (2008) PLoS Comput Biol. Jul 4;4(7):e1000107.
View the abstract, the full text version at PLoS Compu:
Full text.
|

