Usage instructions
1. Specify the training sequences
All the input sequences must be in one-letter amino acid
code. The allowed alphabet (not case sensitive) is as follows:
A C D E F G H I K L M N P Q R S T V W Y
The training sequences can be input in the following two ways:
-
Paste a set of sequences, one sequence per line (just the amino acids)
into the upper left window.
Look here to see an example of the format.
-
You can also select a file (in the same format) on your local disk, either
by typing the file name into the lower left window or by browsing the disk.
3. Select evaluation examples (Optional)
The evaluation examples can either be one example per line
(optionally followed by an assigned value:
example.
) or in fasta format
fasta example.
.
3. Customize your run by changing some of the advanced options (Optional)
4. Submit the job
Click on the
"Submit" button. The status of your job (either 'queued'
or 'running') will be displayed and constantly updated until it terminates and
the server output appears in the browser window.
At any time during the wait you may enter your e-mail address and simply leave
the window. Your job will continue; you will be notified by e-mail when it has
terminated. The e-mail message will contain the URL under which the results are
stored; they will remain on the server for 24 hours for you to collect them.
Output format
DESCRIPTION
Example of output is found below. The output is divided into the folowinng sections:
- Description of training data
- Prediction method
- Parameters for training of Gibbs method
- Prediction data
- Evaluation of predictions (if assignments are supplied by user)
- Predictions
This section contain a line "Peptide Start res Motif Prediction (Assign) Sequence"
Peptide: Peptide number
Start res: 1st residue in motif
Motif: Motif found in sequence
Prediction: Prediction score for motif
Assign: Assignment of sequence (if supplied by user)
Sequence: Sequence containing motif
EXAMPLE OUTPUT
Description of training data
Length of motif: 9
Number of training data: 456
Number of positive training examples: 456
Parameters for training of Gibbs method
Clustering using the Henikoff & Henikoff 1/nr method
Weight on prior: 50.000000
Start temperature: 0.150000
End temperature: 0.000100
Number of temperature steps: 10.000000
Using default seed
Number of iterations per train example: 20
Using amino acid background distribution from SWISSPROT
Equal weights on all positions
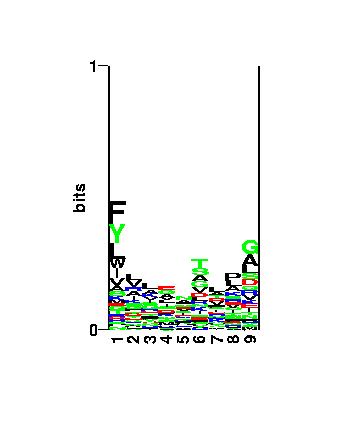
Figure: Visualization of the binding motif using the
logo
program.
A short explanation of HLA supertypes can be found
here.
Alignment generated by Gibbs sampler
Matrix generated by Gibbs sampler
Prediction data
Number of evaluation data: 57
Predicting using a matrix method
Evaluation of predictions
Pearson coefficient for N= 57 data: -0.30553
Aroc value: 0.31622
Threshold for counting example as positive: 50.000000
Predictions
Peptide Start res Motif Prediction Assign Sequence
1 8 FWSFGSEDG 6.549 5.400 MASPGSGFWSFGSEDGSGDS
2 1 FGSEDGSGD 3.304 62.000 FGSEDGSGDSENPGRARAWC
3 6 ARAWCQVAQ 3.363 100.000 ENPGRARAWCQVAQKFTGGI
4 1 QVAQKFTGG 1.638 100.000 QVAQKFTGGIGNKLCALLYG
5 8 LYGDAEKPA 4.603 100.000 GNKLCALLYGDAEKPAESGG
6 7 ESGGSQPPR 1.463 100.000 DAEKPAESGGSQPPRAAARK
7 8 ARKAACACD 3.170 100.000 SQPPRAAARKAACACDQKPC
8 12 CSKVDVNYA -1.160 2.400 AACACDQKPCSCSKVDVNYA
9 12 LHATDLLPA 6.138 0.500 SCSKVDVNYAFLHATDLLPA
10 2 LHATDLLPA 6.138 0.200 FLHATDLLPACDGERPTLAF
11 12 QDVMNILLQ 1.590 100.000 CDGERPTLAFLQDVMNILLQ
12 11 YVVKSFDRS 4.430 0.700 LQDVMNILLQYVVKSFDRST
13 1 YVVKSFDRS 4.430 19.000 YVVKSFDRSTKVIDFHYPNE
14 5 FHYPNELLQ 6.535 5.000 KVIDFHYPNELLQEYNWELA
15 7 WELADQPQN 7.249 5.000 LLQEYNWELADQPQNLEEIL
16 11 MHCQTTLKY 5.006 100.000 DQPQNLEEILMHCQTTLKYA
17 9 YAIKTGHPR 9.089 100.000 MHCQTTLKYAIKTGHPRYFN
18 8 YFNQLSTGL 5.320 0.500 IKTGHPRYFNQLSTGLDMVG
19 12 AADWLTSTA 1.734 1.400 QLSTGLDMVGLAADWLTSTA
20 5 WLTSTANTN 4.697 41.000 LAADWLTSTANTNMFTYEIA
21 5 FTYEIAPVF 3.031 85.000 NTNMFTYEIAPVFVLLEYVT
22 4 MREIIGWPG 7.542 48.000 LKKMREIIGWPGGSGDGIFS
23 9 FSPGGAISN 0.479 80.000 PGGSGDGIFSPGGAISNMYA
24 9 YAMMIARFK 3.854 100.000 PGGAISNMYAMMIARFKMFP
25 11 EVKEKGMAA 3.955 25.000 MMIARFKMFPEVKEKGMAAL
26 4 EKGMAALPR 4.336 40.000 EVKEKGMAALPRLIAFTSEH
27 6 FTSEHSHFS 8.330 0.200 PRLIAFTSEHSHFSLKKGAA
28 3 FSLKKGAAA 5.693 100.000 SHFSLKKGAAALGIGTDSVI
29 10 ILIKCDERG 1.002 24.000 ALGIGTDSVILIKCDERGKM
30 10 MIPSDLERR -0.681 100.000 LIKCDERGKMIPSDLERRIL
31 8 RILEAKQKG 2.013 38.000 IPSDLERRILEAKQKGFVPF
32 10 FLVSATAGT 5.278 4.000 EAKQKGFVPFLVSATAGTTV
33 11 YGAFDPLLA 6.422 7.000 LVSATAGTTVYGAFDPLLAV
34 1 YGAFDPLLA 6.422 100.000 YGAFDPLLAVADICKKYKIW
35 10 WMHVDAAWG 9.248 2.700 ADICKKYKIWMHVDAAWGGG
36 1 MHVDAAWGG 1.533 43.000 MHVDAAWGGGLLMSRKHKWK
37 9 WKLSGVERA 7.137 0.800 LLMSRKHKWKLSGVERANSV
38 9 SVTWNPHKM 4.403 13.000 LSGVERANSVTWNPHKMMGV
39 5 HKMMGVPLQ 3.161 34.000 TWNPHKMMGVPLQCSALLVR
40 8 LVREEGLMQ 5.674 17.000 PLQCSALLVREEGLMQNCNQ
41 3 GLMQNCNQM 2.757 41.000 EEGLMQNCNQMHASYLFQQD
42 5 YLFQQDKHY 5.432 22.000 MHASYLFQQDKHYDLSYDTG
43 5 LSYDTGDKA 4.032 31.000 KHYDLSYDTGDKALQCGRHV
44 12 VFKLWLMWR 0.431 100.000 DKALQCGRHVDVFKLWLMWR
45 5 LWLMWRAKG 8.337 33.000 DVFKLWLMWRAKGTTGFEAH
46 7 FEAHVDKCL 2.003 100.000 AKGTTGFEAHVDKCLELAEY
47 12 YNIIKNREG 3.638 34.000 VDKCLELAEYLYNIIKNREG
48 2 YNIIKNREG 3.638 4.000 LYNIIKNREGYEMVFDGKPQ
49 2 EMVFDGKPQ 2.819 67.000 YEMVFDGKPQHTNVCFWYIP
50 7 WYIPPSLRT 5.691 0.600 HTNVCFWYIPPSLRTLEDNE
51 3 LRTLEDNEE 0.154 5.000 PSLRTLEDNEERMSRLSKVA
52 4 SRLSKVAPV 3.410 100.000 ERMSRLSKVAPVIKARMMEY
53 10 YGTTMVSYQ 2.533 10.000 PVIKARMMEYGTTMVSYQPL
54 3 TMVSYQPLG 1.012 10.000 GTTMVSYQPLGDKVNFFRMV
55 7 FRMVISNPA 11.762 0.700 GDKVNFFRMVISNPAATHQD
56 2 SNPAATHQD 3.258 65.000 ISNPAATHQDIDFLIEEIER
57 8 FLIEEIERL 3.306 20.000 ATHQDIDFLIEEIERLGQDL
Article Abstract
REFERENCE
Improved prediction of MHC class I and class II epitopes using a novel Gibbs sampling approach.
Nielsen M, Lundegaard C, Worning P, Hvid CS, Lamberth K, Buus S, Brunak S, Lund O.
Bioinformatics. 2004 20:1388-97
ABSTRACT
MOTIVATION: Prediction of which peptides will bind a specific major histocompatibility complex (MHC) constitutes an
important step in identifying potential T-cell epitopes suitable as vaccine candidates. MHC class II binding peptides have a
broad length distribution complicating such predictions. Thus, identifying the correct alignment is a crucial part of identifying
he core of an MHC class II binding motif. In this context, we wish to describe a novel Gibbs motif sampler method ideally
suited for recognizing such weak sequence motifs. The method is based on the Gibbs sampling method, and it incorporates
novel features optimized for the task of recognizing the binding motif of MHC classes I and II. The method locates the binding
motif in a set of sequences and characterizes the motif in terms of a weight-matrix. Subsequently, the weight-matrix can be
applied to identifying effectively potential MHC binding peptides and to guiding the process of rational vaccine design.
RESULTS: We apply the motif sampler method to the complex problem of MHC class II binding. The input to the method is
amino acid peptide sequences extracted from the public databases of SYFPEITHI and MHCPEP and known to bind to the MHC
class II complex HLA-DR4(B1*0401). Prior identification of information-rich (anchor) positions in the binding motif is
shown to improve the predictive performance of the Gibbs sampler. Similarly, a consensus solution obtained from an ensemble
average over suboptimal solutions is shown to outperform the use of a single optimal solution. In a large-scale benchmark
calculation, the performance is quantified using relative operating characteristics curve (ROC) plots and we make a detailed
comparison of the performance with that of both the TEPITOPE method and a weight-matrix derived using the conventional
alignment algorithm of ClustalW. The calculation demonstrates that the predictive performance of the Gibbs sampler is higher
than that of ClustalW and in most cases also higher than that of the TEPITOPE method.