Paste a set of peptides (up to 50 amino acids in length), one sequence per line into the upper left window, or upload a
file from your local disk.
The input file can be a plain list of peptides (
Sample 1) or an annotated list (
Sample 2). The annotation is carried over to the results, and may be useful for correlating a known classification with the clustering produced by the method.
All input sequences must be in one-letter amino acid code. The allowed alphabet
(not case sensitive) is as follows:
A C D E F G H I K L M N P Q R S T V W Y
The options are divided in three levels:
Basic,
Filtering and
SOM. The only essential parameters to be set are in the Basic options; if you are using the server for the first time you may leave all the other options unchanged.
A brief explanation of each option can be visualized by hovering the mouse over the

symbol next to each option in the submission form.
BASIC options
Job name:
This prefix is pre-pended to all files generated by the current run. If left empty, a
system-generated number will be assigned as prefix.
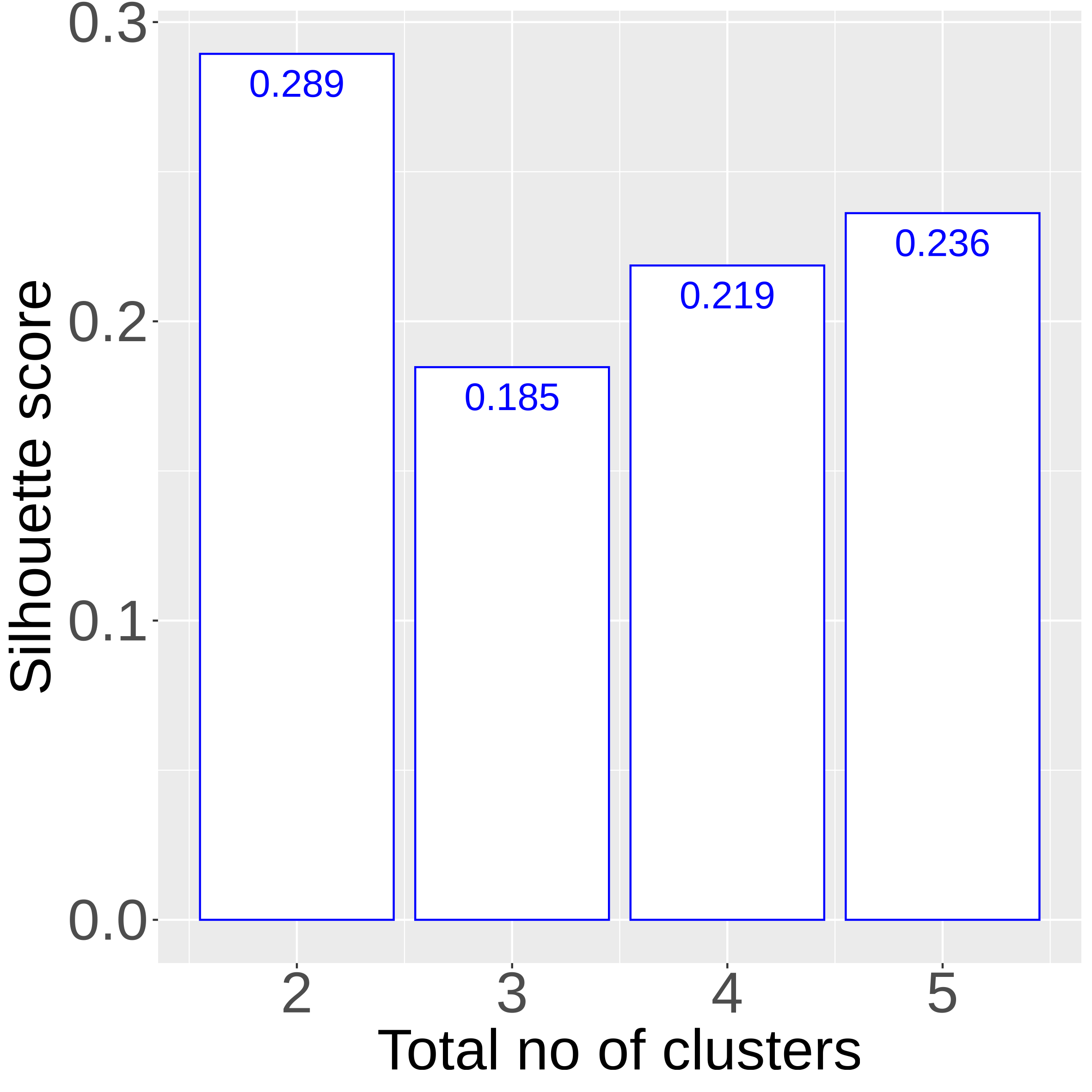
Number of clusters:
You may provide a specific number of clusters (e.g. 3), or an interval of partitions (e.g. 2-5).
In the second case, the method will suggest the optimal number of cluster it found in the
data, given the parameter configuration of the job. Maximum number of clusters: 15.
FILTERING options
Make clustering moves at each iteration:
By default, simple shift moves are performed at each iteration, indel moves every 10
iterations, single peptide moves every 20 iterations, phase shift moves every 100 iterations.
You can alter this behavior by ticking this option; simple shift and phase shift moves
become disabled, and single peptide moves are made at each iteration. This set-up is
recommended for "nearly-aligned" data, where clustering and indels should be sampled more
regularly than extensions at the termini. That is the case, for example, of sets of MHC
class I ligands of different length, which would in most cases require central indels
to model peptide bulging of long ligand.
SOM options
Number of iterations per sequence per temperature step:
This parameter ("I") specificies how long your clustering schedule should be. Note that total
number of iterations is the results of "I" multiplied by the number of sequences times the
number of temperature steps, and it will increase linearly the execution time.
Initial Monte Carlo temperature:
The temperature is a scalar, lowered by discreet steps as the iterations progress. The
temperature influences the probability of accepting or rejecting the moves of the algorith.
In the initial iterations (high temperature) the program is free to explore the landscape
of solutions, and as the system cools off only moves that increase the energy will be
accepted.
Number of temperature steps:
The number of steps in the cooling schedule (starting from the initial temperature specified
above).
Interval between Indel moves:
Specifies how often to attempt introducing insertions and deletions (see glossary).
Interval between Single peptide moves:
Specifies how often to attempt moving a sequence between clusters (see glossary).
Interval between Phase shift moves:
Specifies how often to attempt shifting the alignment window of a single cluster.
Background amino acid frequencies:
Construction of PSSMs relies on calculating the frequency of a given residue at a given position,
compared to the expected background frequency of that amino acid. You may use a flat background model
identical for all amino acids (Flat), a pre-calculated distribution reflecting the relative
frequency of each residue in naturally occurring proteins (Pre-calculated Uniprot), or determine
the background model directly from the dataset you submitted (From data).
Preference for hydrophobic AAs at P1:
In the special case of MHC class II data, we have previously found helpful to guide the
alignment by expressing a preference for hydrophobic residues at the P1 of the alignment.
Sequence weighting type:
Data redundancy may affect the quality of the clustering. You may use an explicit clustering
of the sequences in a given group (Clustering), or use a faster heuristic that calculates the degree of
variability at each column in the alignment (Heuristic, recommended); you may also disable
sequence weighting for downweighting of redundant sequences (None).