Submit data
Mirror
Use DiscoTope-3.0 on BioLib if this server is heavily loaded.
Upload or provide a list of PDB protein structures. For detailed instructions, see the Instructions tab.
Instructions
Instructions
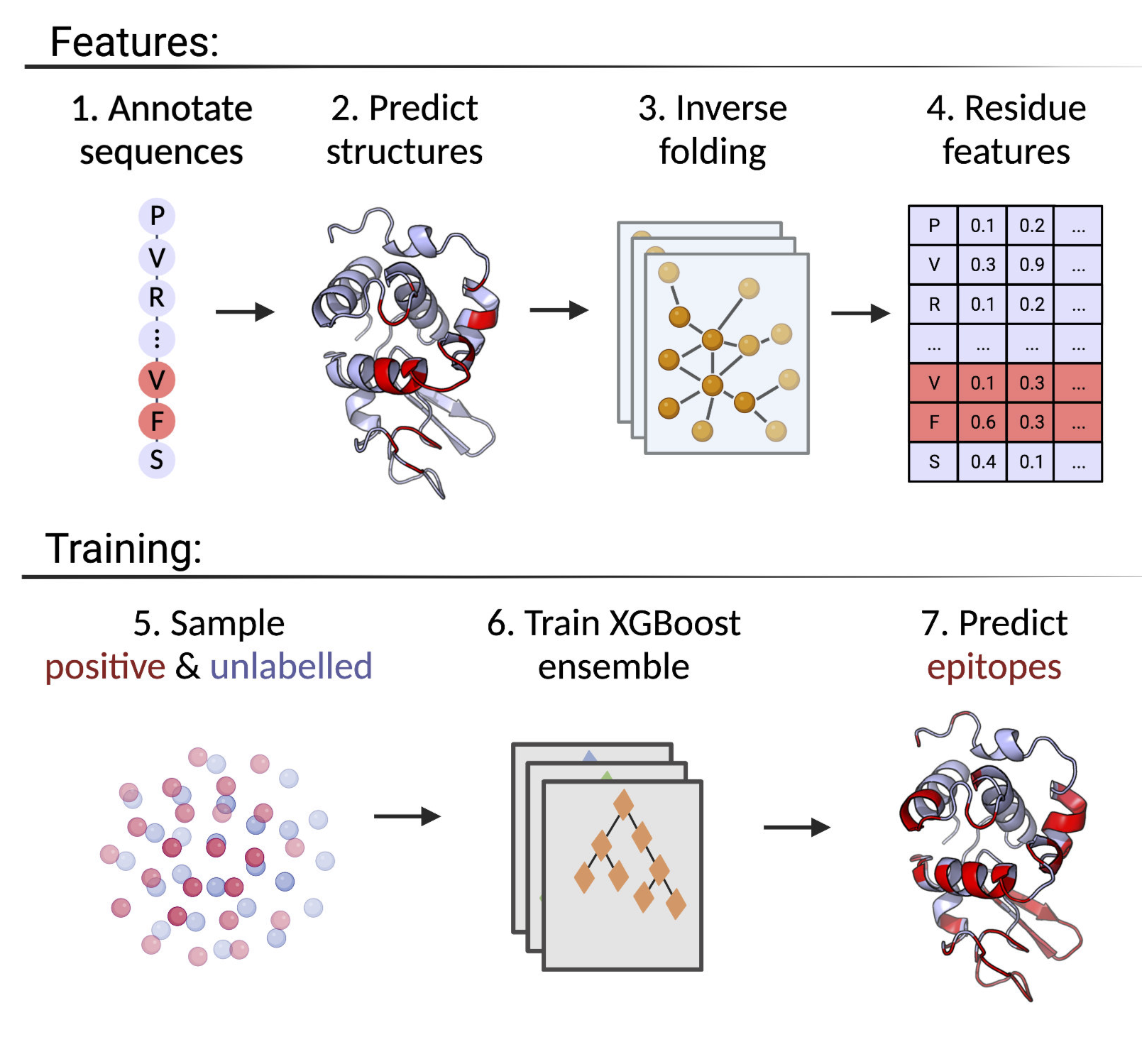
DiscoTope-3.0 predicts per-residue epitope propensity of input protein structures. The server requires input protein structures in the PDB format. These may either be uploaded by the user or downloaded by the DiscoTope-3.0 server.
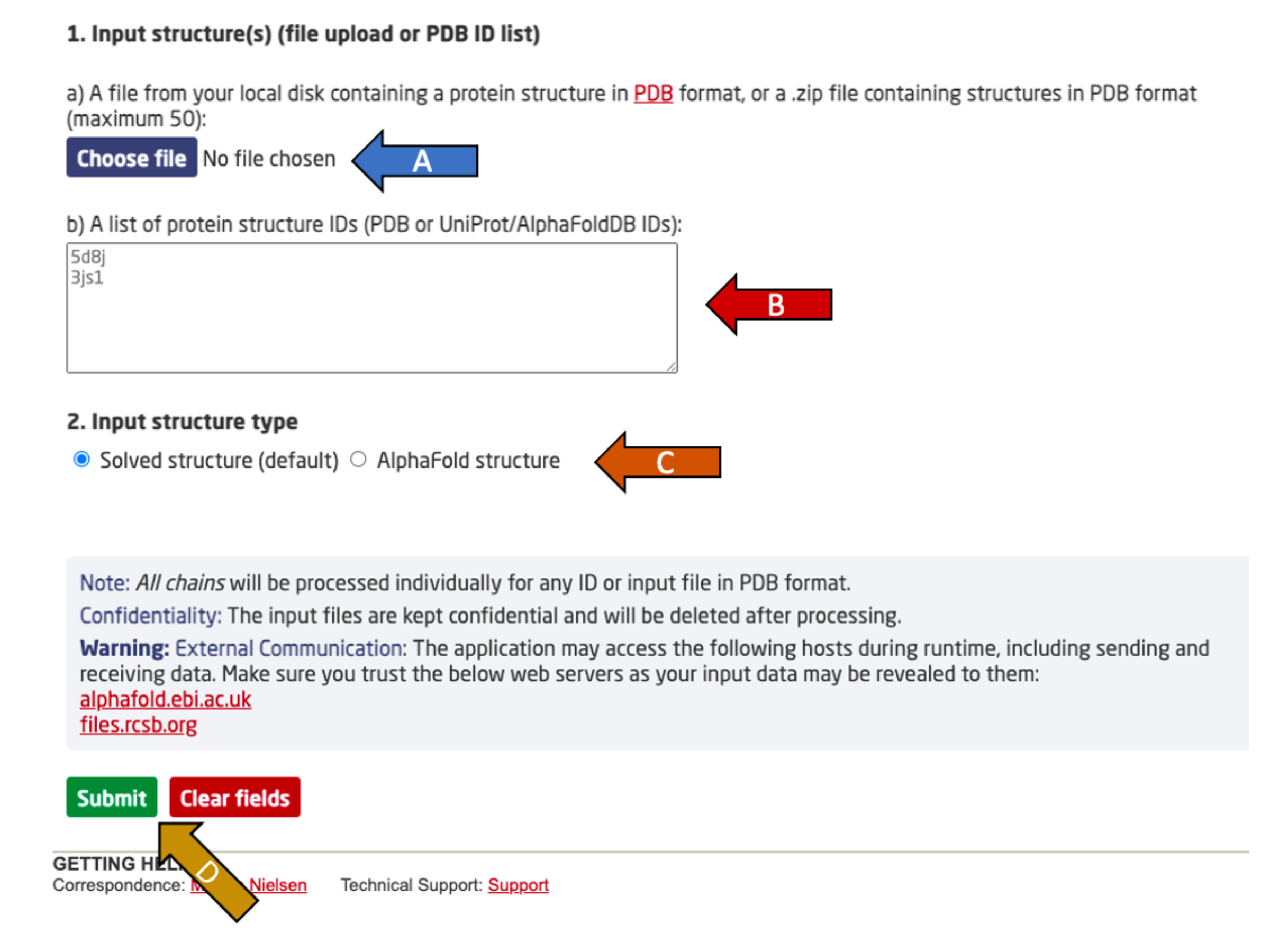
Choose between either:
- Upload a protein PDB structure or a compressed ZIP archive with the Choose file button (arrow A). The ZIP file may contain up to 50 PDBs. The chosen file may not be larger than 30 MB.
- OR
- Enter a list of protein structure IDs, to download from either RCSB or AlphaFoldDB based on the flag set in arrow C. No chain specification should be used (e.g. 5d8j_A). Up to 50 IDs may be given, with one ID per line.
After providing one of the two input options, specify the input structure type (arrow C). Experimental PDB structures (solved) and AlphaFold2 predicted structures are supported.
The suggested calibrated score thresholds correspond to observed epitope percentile scores in the validation set, matching expected recall rates at the given thresholds (see paper).
- Higher confidence (1.50, recall up to ~30 %)
- Moderate confidence (0.90, recall up to ~50 %, default)
- Lower confidence (0.40, recall up to ~70 %)
Finally, click submit to complete the submission (arrow D).
Output format
DiscoTope-3.0 outputs single chain PDB files, with matching per-residue CSV files. These can be visualized on the server with the Mol* viewer.
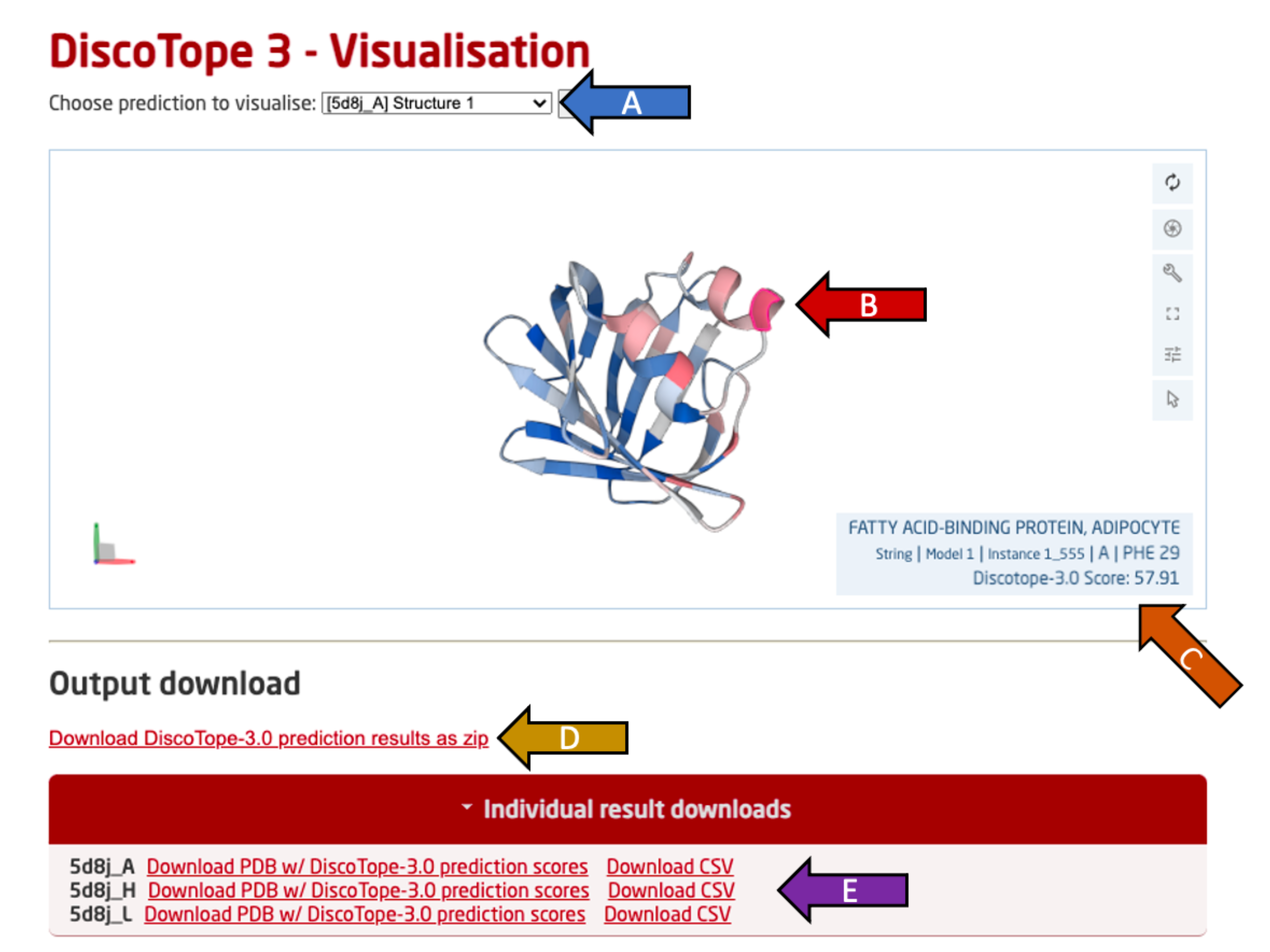
Use the drop-down menu (arrow A) to switch the view to another predicted PDB chain among all provided PDB files.
The selected chain is visualized in an interactive view below using Mol* (arrow B). This view may be rotated freely (click and move), along an axis (hold shift), zoomed (scroll) or moved (hold ctrl) using the mouse and listed keys in parantheses.
Move the cursor over a residue to see it's predicted DiscoTope-3.0 score, residue ID as well as the chain name (arrow C). Residues with higher epitope propensity are colored in a deeper red, while residues with lower epitope propensity are colored in a deeper blue. The color scale is absolute and not adjusted per PDB.
All output predictions may be downloaded as a compressed ZIP archive (arrow D), containing per PDB chain predictions in both .CSV and .PDB format.
Single PDB chain results may be downloaded in a .CSV or .PDB format by clicking the "Individual result downloads" button (arrow E).
The CSV files contains per-residue outputs, with the following column headers:
- PDB ID and chain name
- Chain identifier
- Relative residue index (re-numbered from 1)
- Amino-acid residue, 1-letter
- DiscoTope-3.0 score
- Calibrated DiscoTope-3.0 score, normalized for protein length and surface scores
- Predicted epitope column (based on chosen threshold)
- Relative surface accessibility (Shrake-Rupley, normalized using Sander scale)
- AlphaFold pLDDT score (set to 100 for non-AlphaFold structures)
- Chain length
- A binary feature set to 1 for AlphaFold structures.
The calibrated score thresholds correspond to observed epitope percentile scores in the validation set, matching expected recall rates at the given thresholds (see paper).
- Higher confidence (1.50, recall up to ~30 %)
- Moderate confidence (0.90, recall up to ~50 %, default)
- Lower confidence (0.40, recall up to ~70 %)
If using PyMol, the following commands can be used to visualize the thresholds:
- set_color higher, [0.992, 0.490, 0.302]
- set_color moderate, [0.996, 0.851, 0.212]
- set_color lower, [0.416, 0.796, 0.945]
- set_color very_low, [0.051, 0.341, 0.827]
- color higher, b < 10000
- color moderate, b < 150
- color lower, b < 90
- color very_low, b < 40
Results are displayed using:
Mol* Viewer: modern web app for 3D visualization and analysis of large biomolecular structures
David Sehnal, Sebastian Bittrich, Mandar Deshpande, Radka Svobodová, Karel Berka, Václav Bazgier, Sameer Velankar, Stephen K Burley, Jaroslav Koča, Alexander S Rose.
Nucleic Acids Research (2021). doi: 10.1093/nar/gkab314
Abstract
DiscoTope-3.0: Improved B-cell epitope prediction using inverse folding latent representations
Magnus Haraldson Høie, Frederik Steensgaard Gade, Julie Maria Johansen, Charlotte Würtzen, Ole Winther, Morten Nielsen, Paolo Marcatili.
Frontiers in Immunology (Feb 2024). doi: 10.3389/fimmu.2024.1322712
Accurate computational identification of B-cell epitopes is crucial for the development of vaccines, therapies, and diagnostic tools.
Structure-based prediction methods generally outperform sequence-based models, but are limited by the availability of experimentally solved structures.
Here, we present DiscoTope-3.0, a B-cell epitope prediction tool that exploits inverse folding representations from solved or AlphaFold-predicted structures.
On independent datasets, the method demonstrates improved performance on both linear and non-linear epitopes with respect to current state-of-the-art
algorithms. Most notably, our tool maintains high predictive performance across solved and predicted structures, alleviating the need for experiments and
extending the general applicability of the tool by more than 4 orders of magnitude. DiscoTope-3.0 is available as a web server and downloadable package,
processing up to 50 structures per submission. The web server interfaces with RCSB and AlphaFoldDB, enabling large-scale prediction on all currently
cataloged proteins. DiscoTope-3.0 is available here and on BioLib.