Prediction
of intrinsic aqueous solubility (logS0): The prediction server calculates intrinsic
solubility (solubility of the non-ionized compound) values based on the
chemical structure information. For the prediction a neural network model
trained on a 4548 compound drug-like
data set from the PHYSPROP database, and tested on two different of external
validation sets, is used. The model is built on nine 2D-MOE (Molecular
Operational Environment) descriptors, and the training was done in a three-fold
cross validation using a fully connected feed-forward neural network with one
hidden layer and nine hidden neurons. (The chemical structures can be fed to
the server in SMILES or sdf format, or sketched in
the JME applet, the structures are transformed to 2D sdf-format with the Molconvert
program from ChemAxon and the descriptors at
generated with MOE and fed to the NN-predictor.)
Confidence
estimate for the intrinsic solubility prediction: A simple confidence index is assigned to each predicted solubility
value, indicating how well the compound matches the chemical space of the training
set. Compounds for which the predicted target value or the most important
descriptor fall within two standard deviations of the corresponding values of
the training set are considered to have high accuracy. For compounds that fall
within three standard deviation of either property the prediction accuracy is
evaluated to be moderate, and compounds that fall outside this range are
evaluated to have low prediction accuracy.
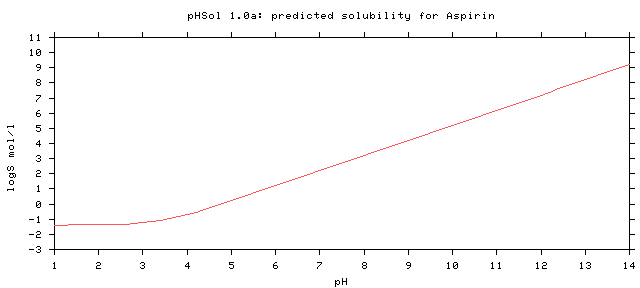
Prediction of pH-dependent aqueous solubility: The predicted pH-solubility profiles (logS) are calculated using the Henderson-Hasselbalch (HH) equation, using the logS0 values predicted with the server described above and acid-base dissociation coefficients (pKa values) computed with the Marvin program from ChemAxon.
In
case of a monoprotic acid the HH equation has the
form
log S = log S0
+ log
(1 + 10pH-pKa)
in the case of a monoprotic base becomes
log S = log S0
+ log
(1 + 10pKa-pH)
and for an ampholyte, the above two equations are combined to give
log S = log S0 + log (1 + 10pH-pKa(acid)
+ 10pKa(base)-pH)
The salt solubility limit of the compounds are not implemented in the present model, but will be included in the next version, and for this purpose a new data set has been measured by colleges at Warsaw University of Technology in Poland.
EXAMPLE OUTPUT
# Compound p_logS0 p_pKa p_pKb Rel. # ======================================================================== Aspirin -1.389 3.41 high # ========================================================================
Aspirin
Download the numerical data