1. Specify the input molecules
This web-server calculates
intrinsic solubility and pH-dependent solubility profiles of drugs and
drug-like molecules from molecular structure.
There are three possible ways of entering molecular structure information.
1) Insert the SMILES strings of the molecules in the window to the left.
2) Read the SMILES strings from a file (*.smi)
3) Read the structures from a sdf file (*.sdf)
Sample copy
The SMILES entry window and the smi file should have the following format (SMILES-string identifier). Multiple structures can be entered on separate lines.
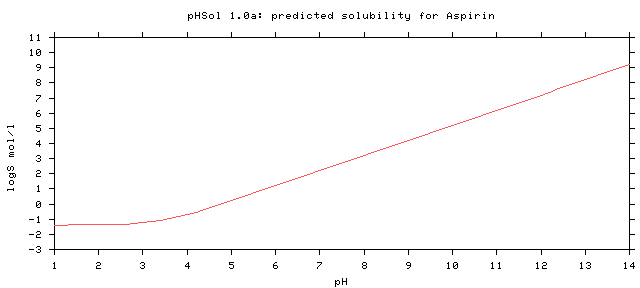
CC(=O)OC1=CC=CC=C1C(=O)O Aspirin
CCOC(=O)C1=CC=C(C=C1)N Benzocaine
C1C2C(C(C1Cl)Cl)C3(C(=C(C2(C3(Cl)Cl)Cl)Cl)Cl)Cl Chlordane
CCOC(=O)CC(C(=O)OCC)SP(=S)(OC)OC Malathion
C1=CC=C(C=C1)C2(C(=O)NC(=O)N2)C3=CC=CC=C3 Phenytoin
CC12CCC3C(C1CCC2O)CCC4=CC(=O)CCC34C Testosterone
Identifiers can be left out, entering SMILES only on separate lines.
2. Customize your run
Click on the button labelled
"Intrinsic solubilities only" if you only want to calculate the intrinsic solubilities.
Click on the button labelled
"Generate graphics" if you want to calculate the pH-dependent solubility profiles as well.
3. Submit the job
Click on the
"Submit" button. The status of your job (either 'queued'
or 'running') will be displayed and constantly updated until it terminates and
the server output appears in the browser window.
At any time during the wait you may enter your e-mail address and simply leave
the window. Your job will continue; you will be notified by e-mail when it has
terminated. The e-mail message will contain the URL under which the results are
stored; they will remain on the server for 24 hours for you to collect them.