DESCRIPTION OF THE SCORES
The scores and graphical output is almost identical to the output
of the SignalP server. The presented scores are calculated in the
same way as for SignalP.
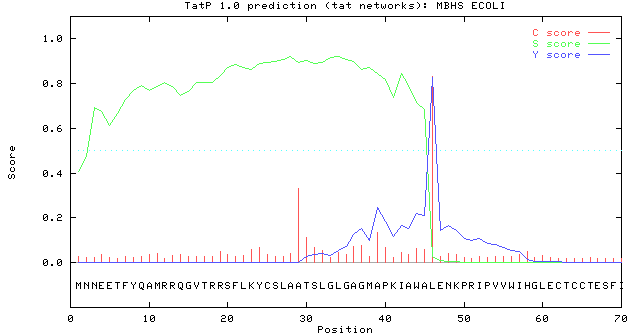
The graphical output from TatP (neural network) comprises three different
scores, C, S and Y. Two additional scores are
reported in the SignalP3-NN output, namely the S-mean and the
D-score, but these are only reported as numerical values.
For each prediction, two different neural networks are used, one for
predicting the actual signal peptide and one for predicting the
position of the signal peptidase I (SPase I) cleavage site. The
S-score for the signal peptide prediction is reported for
every single amino acid position in the submitted sequence, with
high scores indicating that the corresponding amino acid is part
of a signal peptide, and low scores indicating that the amino acid
is part of a mature protein.
The C-score is the ``cleavage site'' score. For each
position in the submitted sequence, a C-score is reported, which
should only be significantly high at the cleavage site. Confusion
is often seen with the position numbering of the cleavage site.
When a cleavage site position is referred to by a single number,
the number indicates the first residue in the mature protein,
meaning that a reported cleavage site between amino acid 26-27
corresponds to that the mature protein starts at (and include)
position 27.
Y-max is a derivative of the C-score combined with the
S-score resulting in a better cleavage site prediction than the
raw C-score alone. This is due to the fact that multiple
high-peaking C-scores can be found in one sequence, where only one
is the true cleavage site. The cleavage site is assigned from the
Y-score where the slope of the S-score is steep and a significant
C-score is found.
The S-mean is the average of the S-score, ranging from the
N-terminal amino acid to the amino acid assigned with the highest
Y-max score, thus the S-mean score is calculated for the length of
the predicted signal peptide.
The D-score is introduced in SignalP version 3.0 and is a
simple average of the S-mean and Y-max score. The score shows
superior discrimination performance of secretory and non-secretory
proteins to that of the S-mean score which was used in SignalP
version 1 and 2. In TatP the D-score is used for final discrimination
of secretory vs. non-secretory.
For non-secretory proteins all the scores represented in the
TatP-NN output should ideally be very low.