Instructions
1. Specify the input sequences
All the input sequences must be in one-letter amino acid
code. The allowed alphabet (not case sensitive) is as follows:
A C D E F G H I K L M N P Q R S T V W Y and X (unknown)
All the alphabetic symbols not in the allowed alphabet
will be converted to X before processing. All the non-alphabetic
symbols, including white space and digits, will be ignored.
The sequences can be input in the following two ways:
-
Paste a single sequence (just the amino acids) or a number of sequences in
FASTA
format into the upper window of the main server page.
-
Select a FASTA
file on your local disk, either by typing the file name into the lower window
or by browsing the disk.
Both ways can be employed at the same time: all the specified sequences will
be processed. However, there may be not more than 5,000 sequences in one submission. The sequences
may not be longer than 10,000 amino acids.
2. Customize your run
- Organism group:
It is important for performance that you choose the correct organism
group —
Archaea, Eukaryotes, Gram-negative bacteria or Gram-positive bacteria —
since the signal peptides of these three groups are known to differ
from each other.
Gram-positive bacteria correspond to
Actinobacteria and
Firmicutes in the
NCBI Taxonomy.
Gram-negative bacteria are all other
eubacteria, except
Tenericutes (including
Mycoplasma), which seem to lack a type I signal peptidase and
therefore do not have standard signal peptides.
- Output format:
You can choose between four output formats:
- Standard
- Appropriate for most users. Shows one plot and one summary per sequence.
- Short
- Convenient if you submit lots of sequences. Shows only one line of
output per sequence and no graphics.
3. Submit the job
Click on the "Submit" button. The status of your job (either 'queued'
or 'running') will be displayed and constantly updated until it terminates and
the server output appears in the browser window.
At any time during the wait you may enter your e-mail address and simply leave
the window. Your job will continue; you will be notified by e-mail when it has
terminated. The e-mail message will contain the URL under which the results are
stored; they will remain on the server for 24 hours for you to collect them.
Example Outputs
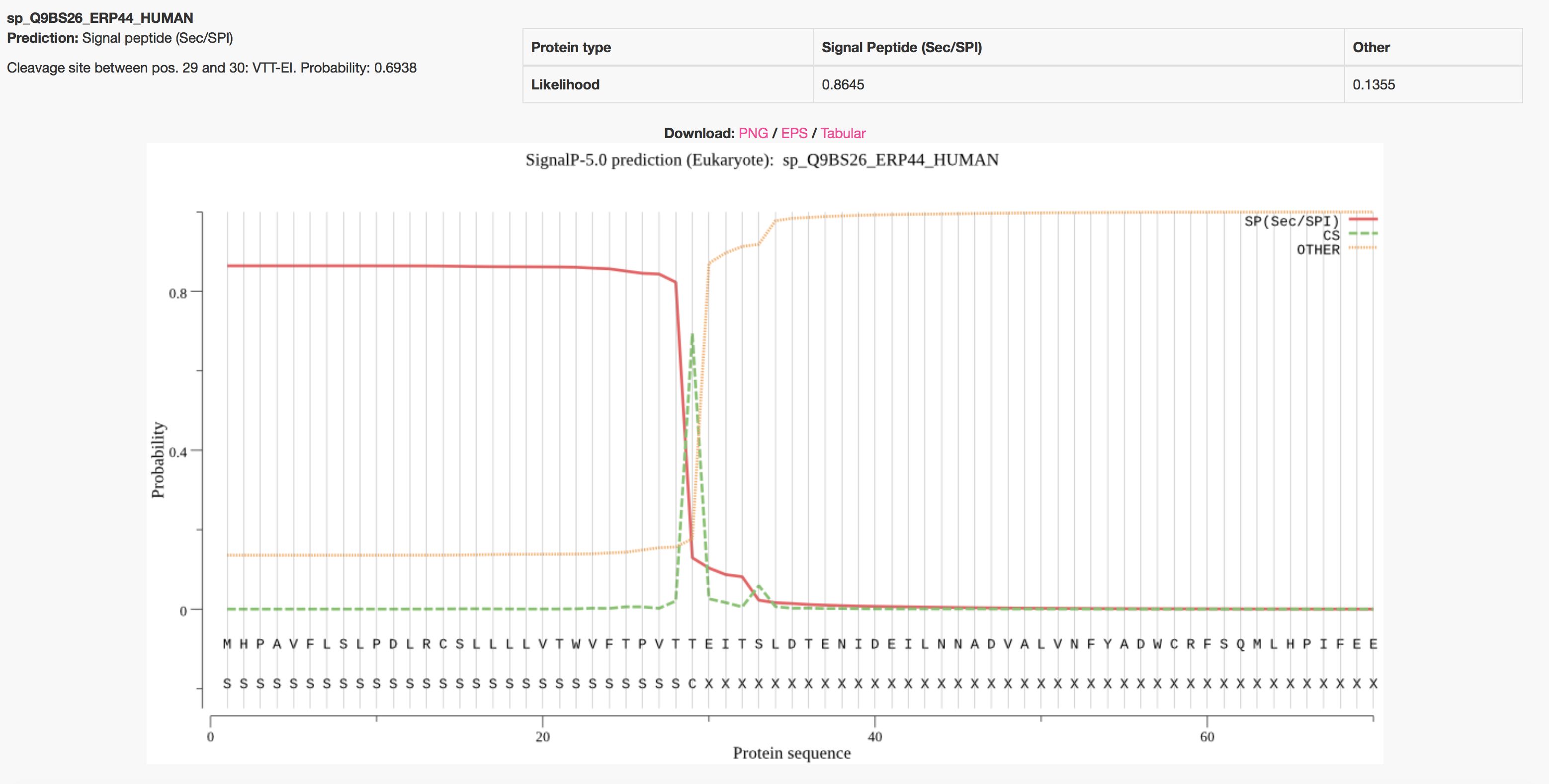
By default the server produces the following output for each input sequence. The example below shows the output for thioredoxin domain containing protein 4 precursor (endoplasmic reticulum protein ERp44), taken from the Uniprot entry ERP44_HUMAN. The signal peptide prediction is consistent with the database annotation.
One annotation is attributed to each protein, the one that has the highest probability. The protein can have a Sec signal peptide (Sec/SPI), a Lipoprotein signal peptide (Sec/SPII), a Tat signal peptide (Tat/SPI) or No signal peptide at all (Other).
If a signal peptide is predicted, the cleavage site position is reported as well.
On the plot, three marginal probabilities are reported, i.e. SP(Sec/SPI) / LIPO(Sec/SPII) / TAT(Tat/SPI) (depending on what type of signal peptide is predicted), CS (the cleavage site) and OTHER (the probability that the sequence does not have any kind of signal peptide).
Example: secretory protein - standard output format
Example: secretory protein - short output format

From the Downloads tab, the user can obtain the results of the run in various formats, i.e. JSON, Prediction summary (results for each submission, 1 line per sequence), Processed entries fasta (a FASTA sequence file containing the sequences of protein that had predicted signal peptides, with the signal peptide removed) and Processed entries gff3 (a file showing the signal peptides feature of those proteins that had predicted signal peptides in GFF3 format).