Performance of SignalP using five-fold cross-validation
SignalP is the most powerful prediction method for signal peptides
published. In order to compare the strength of the neural network
approach to the weight matrix method, we recalculated new weight
matrices from the new data and tested the performance of these. The
weight matrix method was comparable to the neural networks when
calculating C-score, but was practically unable to solve the S-score
problem and therefore did not provide the possibility of calculating
the combined Y-score. The ability to distinguish signal anchors from
signal peptides has not been evaluated for any of the earlier published
methods for signal peptide recognition.
The best prediction of cleavage site location is provided by the
position of the Y-score maximum. The best prediction of sequence
type (signal peptide or non-secretory protein) is given by the mean
S-score (the average of the S-score in the region between position 1
and the position immediately before the Y-score maximum): if mean
S-score is larger than 0.5, the sequence is predicted to be a signal
peptide (see the plot under ``Results: Identification of signal
anchors''). When using these estimates, we obtain the predictive
qualities given in the table below.
These prediction performances are minimal values. They are measured
on the test sets (i.e. data which were not used to train the networks),
and due to the redundancy reduction of the data, the sequence similarity
between training and test sets is so low that the correct cleavage sites
cannot be found by homology. Consequently, the prediction accuracy on
sequences with some degree of homology to the sequences in the data sets
will in general be higher.
| Version
| Cleavage site location
| Signal peptide discrimination
|
| EUK
| Gram-
| Gram+
| EUK
| Gram-
| Gram+
|
| SignalP 1 NN
| 70.2
| 79.3
| 67.9
| 0.97
| 0.88
| 0.96
|
| SignalP 2 NN
| 72.44
| 83.43
| 67.46
| 0.97
| 0.90
| 0.96
|
| SignalP 2 HMM
| 69.51
| 83.43
| 64.50
| 0.94
| 0.93
| 0.96
|
| SignalP 3 NN
| 79.03
| 92.46
| 84.97
| 0.98
| 0.95
| 0.98
|
| SignalP 3 HMM
| 75.70
| 90.22
| 81.58
| 0.94
| 0.94
| 0.98
|
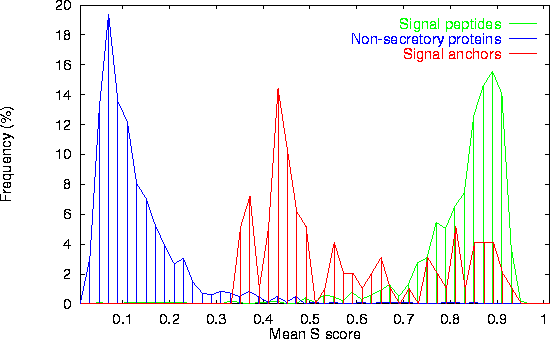
Identification of signal anchors
Above is shown the distribution of the mean S-score for three different
protein types: Signal peptides, Non-secretory proteins (the N-terminal
parts of cytoplasmic or nuclear proteins), and Signal anchors (the
N-terminal parts of type II membrane proteins). Only eukaryotic data
are shown here.
Signal anchors are also referred to as uncleaved signal peptides.
However, they often have sites similar to signal peptide cleavage
sites after their hydrophobic (transmembrane) region. Therefore, a
prediction method can easily be expected to mistake signal anchors for
peptides.
The mean S-score for signal anchors shows some overlap with the signal
peptide distribution (50% of the eukaryotic signal anchor sequences
have mean S-scores larger than 0.5). However, signal anchors are
generally significantly longer than signal peptides. By excluding
signal peptides longer than 35 residues (and using a slightly larger
cutoff), 72% of the eukaryotic signal anchor sequences are correctly
classified. (Only 2.2% of the cleaved eukaryotic signal peptides in
our data set are longer than 35 residues).