Submission
Sequence submission: paste the sequence(s) and/or upload a local file
CITATIONS
For publication of results, please cite as follows. When using predictions
for mammalian sequences:
Feature based prediction of non-classical and leaderless protein secretion
J. Dyrløv Bendtsen, L. Juhl Jensen, N. Blom, G. von Heijne
and S. Brunak
Protein Eng. Des. Sel., 17(4):349-356, 2004
When using predictions for bacterial sequences:
Non-classical protein secretion in bacteria
J. Dyrløv Bendtsen, L. Kiemer, A. Fausbøll and S. Brunak
BMC Microbiology, 5:58, 2005
Instructions
1. Specify the input sequences
All the input sequences must be in one-letter amino acid
code. The allowed alphabet (not case sensitive) is as follows:
A C D E F G H I K L M N P Q R S T V W Y
Please note that the sequences containing other symbols e.g. X
(unknown) will be discarded before processing. The sequences can be input in
the following two ways:
-
Paste a single sequence (just the amino acids) or a number of sequences in
FASTA
format into the upper window of the main server page.
-
Select a FASTA
file on your local disk, either by typing the file name into the lower window
or by browsing the disk.
Both ways can be employed at the same time: all the specified sequences will
be processed. However, there may be not more than 100 sequences and 200,000 amino acids
in total in one submission. The sequences shorter than 15
or longer than 4000 amino acids will be ignored.
2. Select organism
Different prediction models are used for different organisms. You are therefore required to select the organism type prior to running the prediction.
3. Submit the job
Click on the
"Submit" button. The status of your job (either 'queued'
or 'running') will be displayed and constantly updated until it terminates and
the server output appears in the browser window.
At any time during the wait you may enter your e-mail address and simply leave
the window. Your job will continue; you will be notified by e-mail when it has
terminated. The e-mail message will contain the URL under which the results are
stored; they will remain on the server for 24 hours for you to collect them.
NOTE:
SecretomeP is dependent on a number of other programs that have to be run
on the input sequences prior to employing the SecretomeP method itself.
Therefore, the processing of multiple sequences may be time-consuming.
In the case of prolonged wait the user is advised to use the e-mail option
mentioned above.
Output format
DESCRIPTION
For each input sequence the server predicts the possibility of
non-classical secretion.
For eukaryotic sequences, an example of the output is shown below, but it is different in the version for bacteria.
For that version, four scores are generated by the SecretomeP server for each input sequence. The determining score is the 'SecP score', for which a value above 0.5 indicates possible secretion.
This score is the score mentioned in the reference
paper, while the others are provided merely to give an idea about the degree of consent among the three different models used for the prediciton.
Even though SecretomeP is trained to predict
non-classical secretion, it usually gives high score
to proteins entering the classical secretory pathway (having signal peptides). The user is advised to check for conventional signal peptides if in doubt (See SignalP for more information).
EXAMPLE OUTPUT
# Name NN-score Odds Weighted Warning
# by prior
# ============================================================================
FGF1_HUMAN 0.847 4.267 0.009 -
FGF4_HUMAN 0.945 6.804 0.014 signal peptide predicted by SignalP
ATRX_HUMAN 0.093 0.205 0.000 -
In the example above the first protein, known to enter the non-classical
secretory pathway,
FGF1_HUMAN,
is correctly predicted as secretory. The second classical secreted protein,
FGF4_HUMAN,
is correctly predicted as secretory, but a warning that a signal peptide
is predicted is reported. The third protein,
ATRX_HUMAN,
a known nuclear protein, receives a low score, thus is correctly classified.
References
REFERENCE (prediction of non-classical secretion in bacteria)
Non-classical protein secretion in bacteria
J. D. Bendtsen, L. Kiemer, A. Fausbøll and S. Brunak.
BMC Microbiology 2005, 5:58
ABSTRACT
Background: We present an overview of bacterial non-classical
secretion and a prediction method for identification of proteins
following signal peptide independent secretion pathways. We have
compiled a list of proteins found extracellularly despite the
absence of a signal peptide. Some of these proteins also have
known roles in the cytoplasm, which means they could be so-called
``moon-lightning'' proteins having more than one function.
Methods: A thorough literature search was conducted to compile a list of
currently known bacterial non-classically secreted proteins.
Pattern finding methods were applied to the sequences in order to
identify putative signal sequences or motifs responsible for their
secretion. Finally, artificial neural networks were used to
construct protein feature based methods for identification of
non-classically secreted proteins in both Gram-positive and
Gram-negative bacteria.
Results: We have found no signal or motif characteristic to any majority of the proteins
in the compiled list of non-classically secreted proteins, and
conclude that these proteins, indeed, seem to be secreted in a
novel fashion. However, we also show that the apparently
non-classically secreted proteins are still distinguished from
cellular proteins by properties such as amino acid composition,
secondary structure and disordered regions. Specifically,
prediction of disorder reveals that bacterial secretory proteins
are more structurally disordered than their cytoplasmic
counterparts.
Conclusions: We present a publicly
available prediction method capable of discriminating between this
group of proteins and other proteins, thus allowing for the
identification of novel non-classically secreted proteins. We
suggest candidates for non-classically secreted proteins in
Escherichia coli and Bacillus subtilis. The
prediction method is available at
http://www.cbs.dtu.dk/services/SecretomeP-2.0/.
PMID: 16212653
doi: 10.1186/1471-2180-5-58
Supplementary material
Prokaryotic proteome scans
E. coli
Download file with top scoring proteins from E. coli excluding signal peptide containing sequences.
B. subtilis
Download file with top scoring proteins from B. subtilis excluding signal peptide containing sequences.
Performance of the method
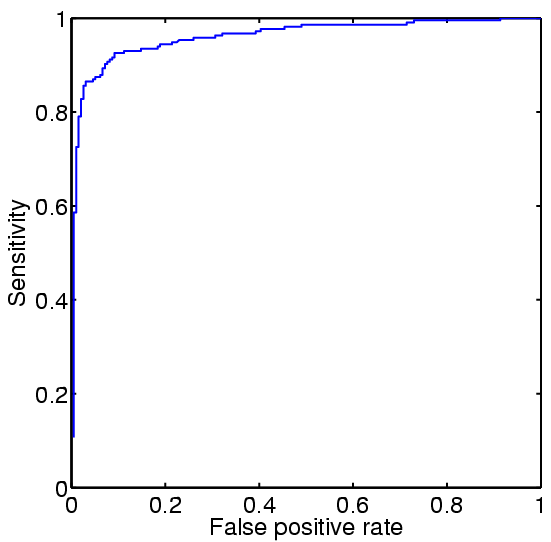
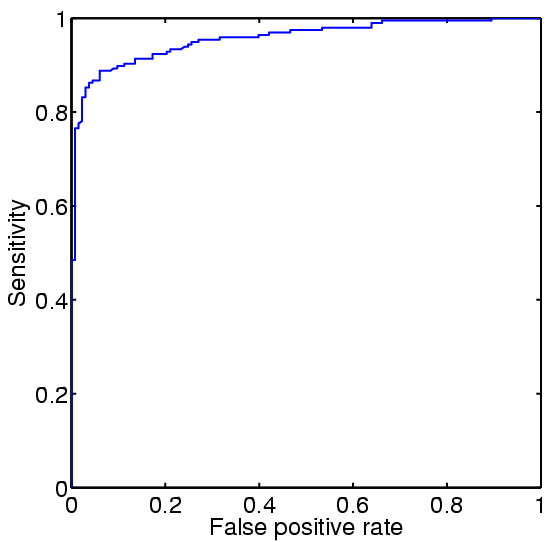
ROC curves for Gram-negative and Gram-positive, respectively.
Features for Gram-