NetSurfP-3.0 web server
NetSurfP-3.0: accurate and fast prediction of protein structural features by protein language models and deep learning
Magnus Haraldson Høie, Erik Nikolas Kiehl, Bent Petersen, Henrik Nielsen, Ole Winther, Jeppe Hallgren, Paolo Marcatili.
Nucleic Acid Research (June 2022). doi: 10.1093/nar/gkac439
Recent advances in machine learning and natural language processing have made it possible to profoundly advance our ability to accurately predict protein structures and their functions. While such improvements are significantly impacting the fields of biology and biotechnology at large, such methods have the downside of high demands in terms of computing power and runtime, hampering their applicability to large datasets. Here, we present NetSurfP-3.0, a tool for predicting solvent accessibility, secondary structure, structural disorder and backbone dihedral angles for each residue of an amino acid sequence. This NetSurfP update exploits recent advances in pre-trained protein language models to drastically improve the runtime of its predecessor by two orders of magnitude, while displaying similar prediction performance. We assessed the accuracy of NetSurfP-3.0 on several independent test datasets and found it to consistently produce state-of-the-art predictions for each of its output features, with a runtime that is up to to 600 times faster than the most commonly available methods performing the same tasks. The tool is freely available as a web server with a user-friendly interface to navigate the results, as well as a standalone downloadable package.
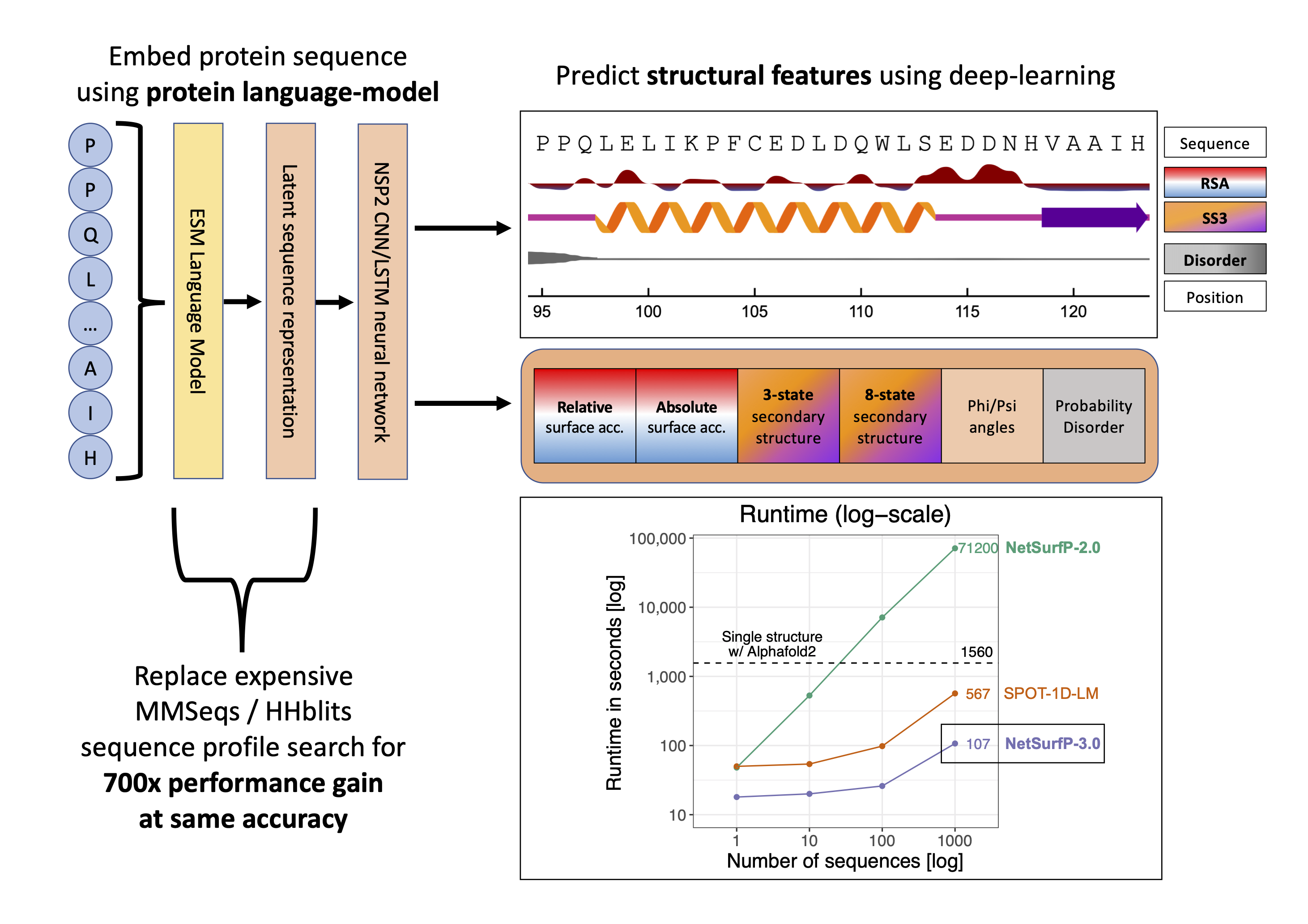
The NetSurfP-3.0 server predicts the following structural features; surface accesibility, secondary structure, disorder, and phi and psi angles using a single model. This NetSurfP update exploits recent advances in pre-trained protein language models to drastically improve the runtime of its predecessor by two orders of magnitude while displaying similar prediction performance.
Instructions
The server requires protein sequence(s) in fasta format, and can not handle nucleic acid sequences.
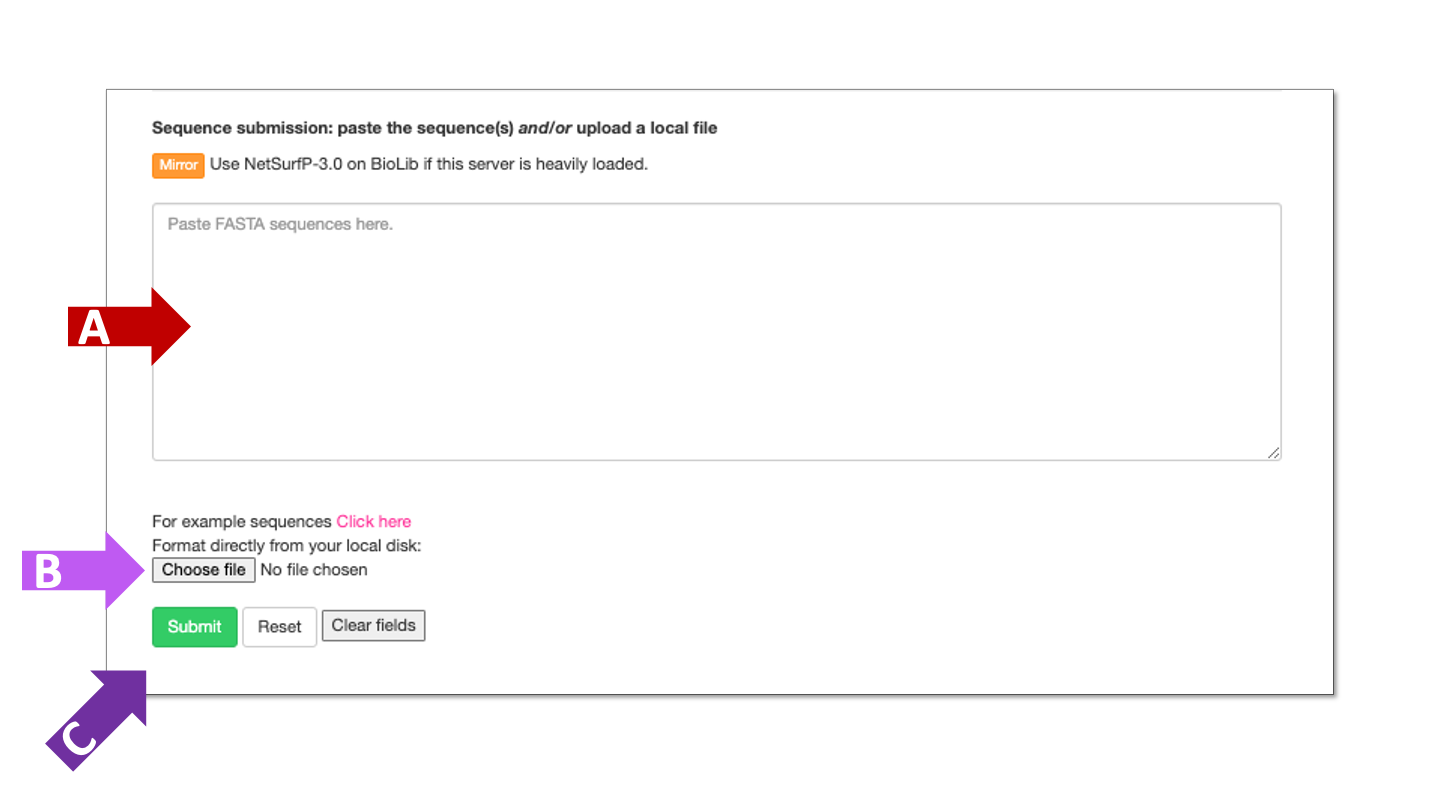
Paste protein sequence(s) in fasta format into field marked by arrow A or upload a fasta file marked by arrow B. Click the submit button, marked by arrow C, when protein sequences are entered.
After the server successfully finishes the job, a Server Output page shows up. If an error happens during prediction a log will appear specifying the error.

Use the navigation bar (arrow A) to flip through the various output pages. The output can be exported in the format of CSV, JSON, NetSurfP-1.0 format and Zipped Archive by clicking "Export All" on the far right of the navigation bar (arrow B). The Zipped Archive contains the NetSurfP-3.0 predictions. The default page is the Server Output, where a short description and summary of the prediction (arrow C), followed by a paginated list of the predicted protein sequences (arrow D). The search bar allows easy accessibility of viewing a specific entry. Each predicted protein can be also be exported separately using the Export button on the far right of the protein of interest.
NetSurfP-3.0 Training/Test Data
Raw data is given in Numpy (Python) compressed files with an array of pdb/chain ids (pdbids) and a 3-dimensional array of input and output features.
First dimension is samples, second dimension is sequence position and third dimension is input features:
# [0:20] Amino Acids (sparse encoding)
# Unknown residues are stored as an all-zero vector
# [20:50] hmm profile
# [50] Seq mask (1 = seq, 0 = empty)
# [51] Disordered mask (0 = disordered, 1 = ordered)
# [52] Evaluation mask (For CB513 dataset, 1 = eval, 0 = ignore)
# [53] ASA (isolated)
# [54] ASA (complexed)
# [55] RSA (isolated)
# [56] RSA (complexed)
# [57:65] Q8 GHIBESTC (Q8 -> Q3: HHHEECCC)
# [65:67] Phi+Psi
# [67] ASA_max