DTU Health Tech
Department of Health Technology
DTU Health Tech
Department of Health Technology
Server predicts the surface accessibility, secondary structure, disorder, and phi/psi dihedral angles of amino acids in an amino acid sequence.
There has been some portability issues for the output. This will be taken care for later. It is possible to see and export the output. Notice: it is a slow service.
| NOTE: This is not the newest version of NetSurfP. To use the current version, please go to the main NetSurfP site! |
Michael Schantz Klausen, Martin Closter Jespersen, Henrik Nielsen, Kamilla Kjærgaard Jensen, Vanessa Isabell Jurtz, Casper Kaae Sønderby, Morten Otto Alexander Sommer, Ole Winther, Morten Nielsen, Bent Petersen, and Paolo Marcatili. NetSurfP-2.0: Improved prediction of protein structural features by integrated deep learning . Proteins: Structure, Function, and Bioinformatics (Feb. 2019). doi: 10.1002/prot.25674
The ability to predict a protein’s local structural features from the primary sequence is of paramount importance for unraveling its function if no solved structures of the protein or its homologs are available. Here we present NetSurfP-2.0 ( https://services.healthtech.dtu.dk/service.php?NetSurfP-2.0 ), an updated and extended version of the tool that can predict the most important local structural features with unprecedented accuracy and run-time. NetSurfP-2.0 is sequence-based and uses an architecture composed of convolutional and long short-term memory neural networks trained on solved protein structures. Using a single integrated model, NetSurfP-2.0 predicts solvent accessibility, secondary structure, structural disorder, and backbone dihedral angles for each residue of the input sequences.
We assessed the accuracy of NetSurfP-2.0 on several independent validation datasets and found it to consistently produce state-of-the-art predictions for each of its output features, with a significant improvement for solvent accessibility and disorder. In addition to improved prediction accuracy the processing time has been optimized to allow proteome wide predictions of more than 4,000 proteins in less than 10 hours.
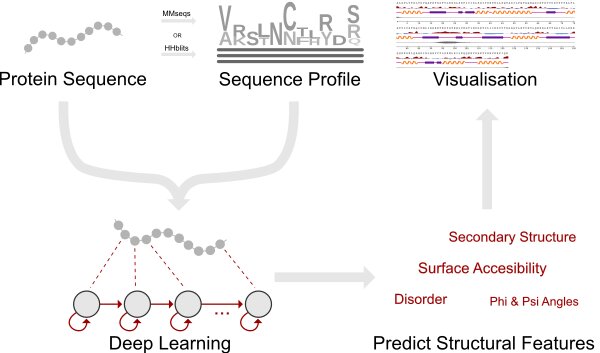
The NetSurfP-2.0 server predicts the following structural features; surface accesibility, secondary structure, disorder, and phi and psi angles. A single model, using a combination of Convolutional and Bi-Directional Long-Short Term Memory Neural Networks, predicts all structural features together.
The server requires protein sequence(s) in fasta format, and can not handle nucleic acid sequences.

Paste protein sequence(s) in fasta format into field marked by arrow A or upload a fasta file marked by arrow B . Click the submit button, marked by arrow C , when protein sequences are entered.
Depending on the sample size, MMseqs (batch) or HHblits (single) will automatically be selected to optimise computational time. Computational time can range from a couple of minutes to several hours depending on the queue and the sample size.
After the server successfully finishes the job, a Server Output page shows up. If an error happens during prediction a log will appear specifying the error.

Use the navigation bar ( arrow A ) to flip through the various output pages. The output can be exported in the format of CSV, JSON, NetSurfP-1.0 format and Zipped Archive by clicking "Export All" on the far right of the navigation bar ( arrow B ). The Zipped Archive contains the sequence profiles from HHblits/MMseqs and the NetSurfP-2.0 predictions. The default page is the Server Output, where a short description and summary of the prediction ( arrow C ), followed by a paginated list of the predicted protein sequences ( arrow D ). The search bar allows easy accessibility of viewing a specific entry. Each predicted protein can be also be exported separately using the Export button on the far right of the protein of interest.
Raw data is given in Numpy (Python) compressed files with an array of pdb/chain ids (pdbids) and a 3-dimensional array of input and output features.
First dimension is samples, second dimension is sequence position and third dimension is input features:
# [0:20] Amino Acids (sparse encoding) # Unknown residues are stored as an all-zero vector # [20:50] hmm profile # [50] Seq mask (1 = seq, 0 = empty) # [51] Disordered mask (0 = disordered, 1 = ordered) # [52] Evaluation mask (For CB513 dataset, 1 = eval, 0 = ignore) # [53] ASA (isolated) # [54] ASA (complexed) # [55] RSA (isolated) # [56] RSA (complexed) # [57:65] Q8 GHIBESTC (Q8 -> Q3: HHHEECCC) # [65:67] Phi+Psi # [67] ASA_max
If you need help regarding technical issues (e.g. errors or missing results) contact Technical Support. Please include the name of the service and version (e.g. NetPhos-4.0). If the error occurs after the job has started running, please include the JOB ID (the long code that you see while the job is running).
If you have scientific questions (e.g. how the method works or how to interpret results), contact Correspondence.
Correspondence:
Technical Support: