CITATIONS
For publication of results, please cite:
DATA RESOURCES
Data resources used to develop this server was obtained from
- IEDB database.
- Quantitative peptide binding data were obtained
from the IEDB database.
- IMGT/HLA database. Robinson J, Malik A, Parham P, Bodmer JG,
Marsh SGE: IMGT/HLA - a sequence database for the human major histocompatibility complex. Tissue Antigens (2000),
55:280-287.
- HLA protein sequences were obtained from the IMGT/HLA database (version 3.1.0).
INSTRUCTIONS
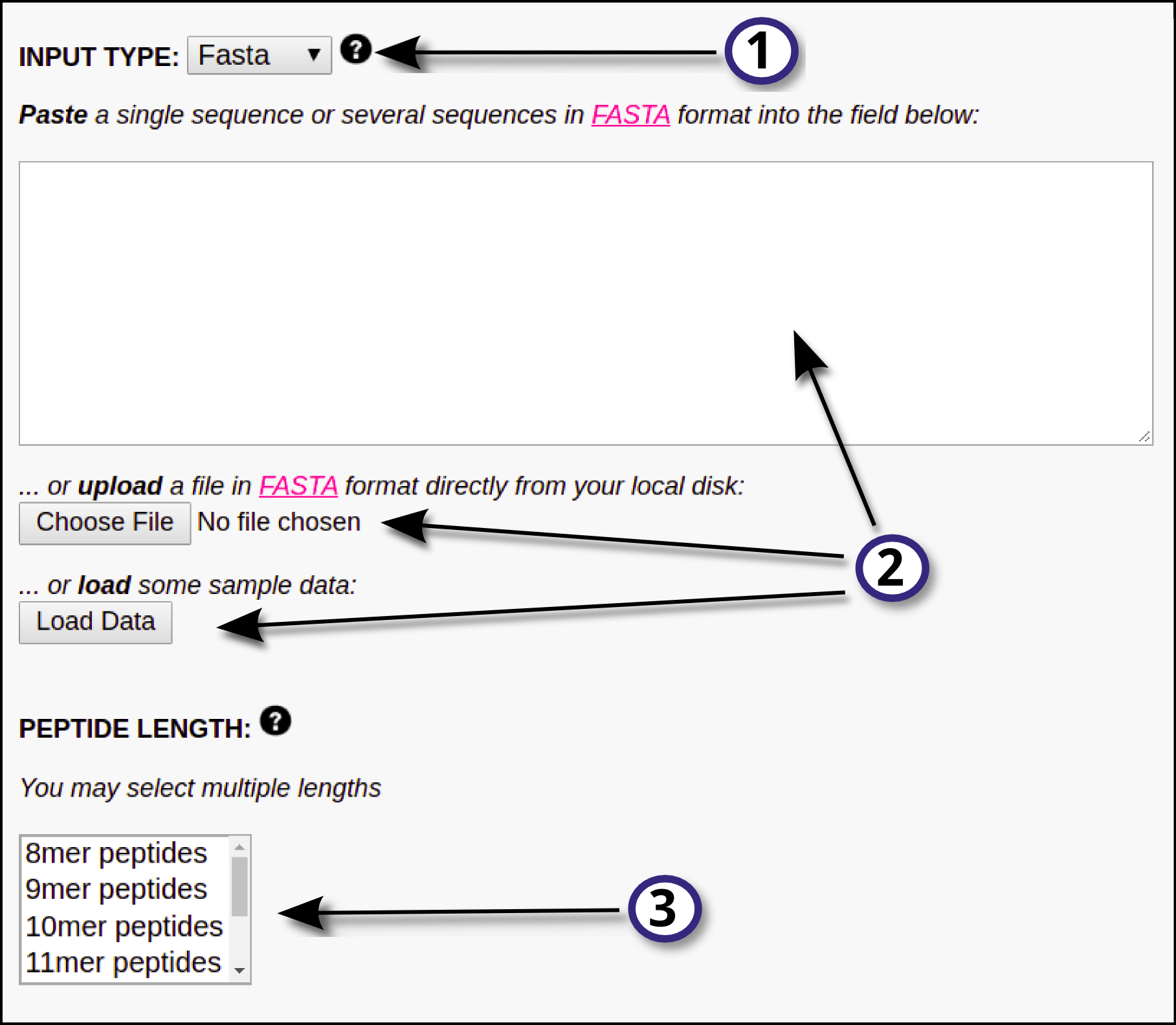
INPUT DATA
In this section, the user must define the input for the prediction server following these steps:
1) Specify the desired type of input data (FASTA or PEPTIDE) using the drop down menu.
2) Provide the input data by means of pasting the data into the blank field, uploading it using the "Choose File" button or by loading sample data using the "Load Data" button. All the input sequences must be in one-letter amino acid code. The alphabet is as follows (case sensitive):
A C D E F G H I K L M N P Q R S T V W Y s t y and X (unknown)
Any other symbol will be converted to X before processing. At most 5000 sequences are allowed per submission; each sequence must be not more than 20,000 amino acids long and not less than 8 amino acids long.
3) If FASTA was selected as input type, the user must select the peptide length(s) the prediction server is going to work with. NetMHCphosPan-1.0 will "chop" the input FASTA sequence in overlapping peptides of the provided length(s) and will predict binding against all of them. By default input proteins are digested into 9-mer peptides. Note that, if PEPTIDE was selected as input type, this step is unnecessary and thus the peptide length selector will directly not appear in the interface.
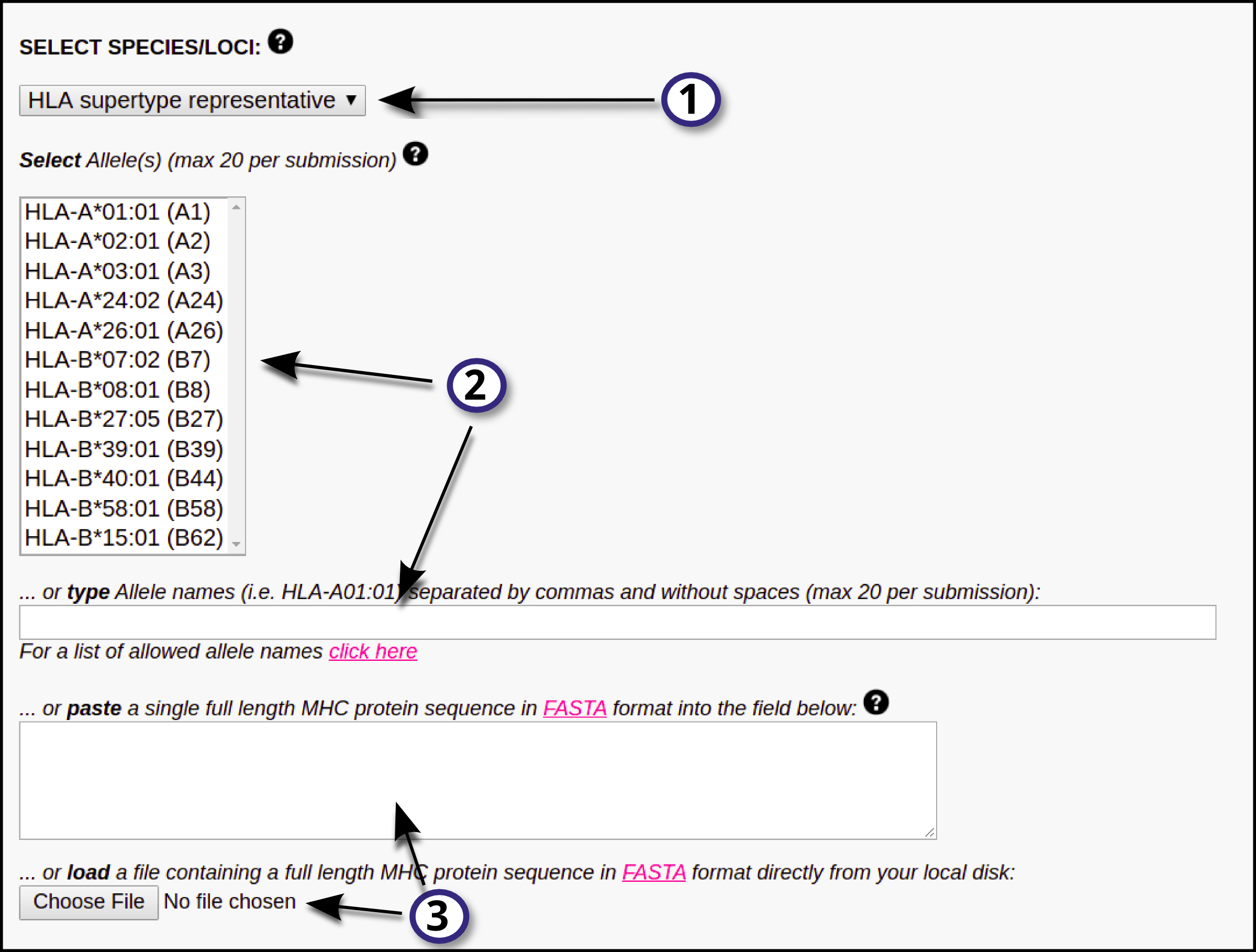
MHC SELECTION
Here, the user must define which MHC(s) molecule(s) the input data is going to be predicted against:
1) First, select the HLA/MHC supertype family.
2) After selecting the MHC family, the user will be able to select a single or multiple MHC molecules from the updated "Select Allele(s)" list. On the other hand, the user may opt to directly type the MHC names in the provided blank field (separated by commas and without blank spaces); if this is the case, there will be no need to select an MHC supertype familiy from the drop-down menu. Click here for a list of MHC molecule names (use the names in the first column). Please note that a maximum of 20 MHC types is allowed per submission.
3) Optionally, the user may choose to paste a full MHC protein sequence in the blank box, or directly upload it by clicking the "Choose file" button. Such sequence must be in FASTA format.
Please note that steps 2) and 3) are mutually exclusive, and are only labeled this way for explanation purposes.
ADDITIONAL CONFIGURATION
In this section, the user may define additional parameters to further customize the run:
1, 2) Specify thresholds for strong and weak binders. They are expressed in terms of %Rank, that is percentile of the predicted binding affinity compared to the distribution of affinities calculated on set of random natural peptides. The peptide will be identified as a strong binder if it is found among the top x% predicted peptides, where x% is the specified threshold for strong binders (by default 0.5%). The peptide will be identified as a weak binder if the % Rank is above the threshold of the strong binders but below the specified threshold for the weak binders (by default 2%).
3) Specify a %Rank threshold to filter out predictions. Only sequences with a predicted %Rank value less than the specified threshold will be printed. To print all predictions, leave this value set to -99.
5) Tick this box to have the output sorted by descending prediction score.
6) Enable this option to export the prediction output to .XLS format (readable by most spreadsheet softwares, like Microsoft Excel). This .XLS file is composed of the following columns:
Three "common" columns:
1. Pos: peptide position (starting from 0).
2. Peptide: peptide sequence.
3. ID: Protein ID.
Six additional columns for each MHC selected for prediction:
4. Core: minimal 9 amino acid binding core (directly in contact with the MHC).
5. Icore: interaction core. This is the sequence of the binding core including eventual insertions or deletions.
6. EL-score: raw eluted ligand likelihood prediction score.
7. EL_Rank: Rank of the predicted eluted ligand likelihood score compared to a set of random natural peptides.
And, finally, two last "common" columns:
10. Ave: average over the raw EL predictions.
11. NB: number of alleles a given peptide binds with EL %RANK threshold < 2%.

SUBMISSION
After the user has finished the "INPUT DATA", "MHC SELECTION" and "ADDITIONAL CONFIGURATION" steps, the submission can now be done. To do so, the user can click on "Submit" to submit the job to the processing server, or click on "Clear fields" to clear the page and start over.
The status of your job (either 'queued' or 'running') will be displayed and constantly updated until it terminates and the server output appears in the browser window.
After the server has finished running the corresponding predictions, an
output page will be delivered to the user. A description of the output format can be found HERE.
At any time during the wait you may enter your e-mail address and simply leave the window. Your job will continue; when it terminates you will be notified by e-mail with a URL to your results. They will be stored on the server for 24 hours.
OUTPUT FORMAT
EXAMPLE
For the following FASTA input example:
>seq1
TMDKsELVQKAKLAEQAERyDDMAAAMKAVtEQGHELsNEERNLLSVAYKNVVGARRSS
WRVISSIEQKTERNEKKQQMGKEYREKIEAELQDICNDVLQLLDKYLIPNATQPESKVF
YLKMKGDYFRYLSEVASGDNKQTTVSNSQQAYQEAFEISKKEMQPTHPIRLGLALNFSV
FYY
With parameters:
Peptide length: 8, 9, 10, 11
Allele: HLA-A*0301
Sort by prediction score: On
NetMHCphosPan-1.0 will return the following output (showing the first 10 predicted peptides):
# NetMHCphosPan version 1.0
# Tmpdir made /usr/opt/www/webface/tmp/server/netmhcphospan/6075C34400004FE4E612AA39/netMHCphospanWhZ7rZ
# Input is in FSA format
# Peptide length 8,9,10,11
# Make EL predictions
HLA-A03:01 : Distance to training data 0.000 (using nearest neighbor HLA-A03:01)
# Rank Threshold for Strong binding peptides 0.500
# Rank Threshold for Weak binding peptides 2.000
------------------------------------------------------------------------------------------------------------
Pos HLA Peptide Core Of Gp Gl Ip Il Icore Identity Score_EL Rnk_EL BindLevel
------------------------------------------------------------------------------------------------------------
1 HLA-A*03:01 TMDKsELVQK TMKsELVQK 0 2 1 0 0 TMDKsELVQK seq1 0.5927991 0.519 <= WB
4 HLA-A*03:01 KsELVQKAK KsELVQKAK 0 0 0 0 0 KsELVQKAK seq1 0.3412235 1.228 <= WB
2 HLA-A*03:01 MDKsELVQK MDKsELVQK 0 0 0 0 0 MDKsELVQK seq1 0.1627113 2.439
1 HLA-A*03:01 TMDKsELVQKA TMKsELVQK 0 2 1 0 0 TMDKsELVQK seq1 0.0528774 4.981
2 HLA-A*03:01 MDKsELVQKAK MDKsELVQK 0 8 2 0 0 MDKsELVQKAK seq1 0.0292524 6.825
26 HLA-A*03:01 AMKAVtEQGH AMKAtEQGH 0 4 1 0 0 AMKAVtEQGH seq1 0.0284383 6.922
29 HLA-A*03:01 AVtEQGHEL AVtEQGHEL 0 0 0 0 0 AVtEQGHEL seq1 0.0238823 7.551
19 HLA-A*03:01 RyDDMAAAMK RyDDMAAMK 0 5 1 0 0 RyDDMAAAMK seq1 0.0203104 8.230
12 HLA-A*03:01 KLAEQAERy KLAEQAERy 0 0 0 0 0 KLAEQAERy seq1 0.0162555 9.162
28 HLA-A*03:01 KAVtEQGHEL KVtEQGHEL 0 1 1 0 0 KAVtEQGHEL seq1 0.0119683 10.560
DESCRIPTION
The prediction output for each molecule consists of the following columns:
Pos: Residue number (starting from 0) of the peptide in the protein sequence.
MHC: Specified MHC molecule / Allele name.
Peptide: Amino acid sequence of the potential ligand.
Core: The minimal 9 amino acid binding core directly in contact with the MHC.
Of: The starting position of the Core within the Peptide (if > 0, the method predicts a N-terminal protrusion).
Gp: Position of the deletion, if any.
Gl: Length of the deletion, if any.
Ip: Position of the insertion, if any.
Il: Length of the insertion, if any.
Icore: Interaction core. This is the sequence of the binding core including eventual insertions of deletions.
Identity: Protein identifier, i.e. the name of the FASTA entry.
Score_EL: The raw prediction score.
%Rank_EL: Rank of the predicted binding score compared to a set of random natural peptides. This measure is not affected by inherent bias of certain molecules towards higher or lower mean predicted affinities. Strong binders are defined as having %rank<0.5, and weak binders with %rank<2. We advise to select candidate binders based on %Rank rather than Score
BindLevel: (SB: Strong Binder, WB: Weak Binder). The peptide will be identified as a strong binder if the %Rank is below the specified threshold for the strong binders (by default, 0.5%). The peptide will be identified as a weak binder if the %Rank is above the threshold of the strong binders but below the specified threshold for the weak binders (by default, 2%).
NOTES
Peptide vs. iCore vs. Core
Three amino acid sequences are reported for each row of predictions:
The Peptide is the complete amino acid sequence evaluated by NetMHCpan. Peptides are the
full sequences submitted as a peptide list, or the result of digestion of source proteins (Fasta submission)
The iCore is a substring of Peptide, encompassing
all residues between P1 and P-omega of the MHC. For all intents and purposes, this is the minimal candidate
ligand/epitope that should be considered for further validation.
The Core is always 9 amino acids long,
and is a construction used for sequence aligment and identification of binding anchors.
ARTICLE ABSTRACTS
MAIN REFERENCE
NetMHCphosPan - Pan-specific prediction of MHC class I antigen presentation of phosphorylated ligands
Carina Thusgaard Refsgaard 1, Carolina Barra 1, Nicola
Ternette, and Morten Nielsen1,2
In preparations, April, 2021
Post-translational modifications of proteins play a crucial part of carcinogenesis. Phosphorylated peptides have shown the ability to be presented by HLA class I molecules and recognised by cytotoxic T cells, making them a promising target for immunotherapy. Identification of MHC class I ligands have so far predominantly been done with bioinformatic tools trained on unmodified peptides. Only one prediction tool has been developed so far and the current tool has been trained only on a limited number of alleles with a limited peptide length coverage (only including 9-mers).
Here we propose a predictor, termed NetMHCphosPan, for HLA presented phospho-peptides. The method is trained using the NNAlign_MA framework, which allows incorporating mixed data types and information leverage between data sets resulting in a greatly improved HLA and peptide length coverage and an overall increased predictive power. Motif deconvolution suggested a strong preference for phosphosites to be located in position 4 of the binding motif, an enrichment of proline at P5 and arginine at P1.
Conducting a large benchmark on data independent from the model development confirmed the superior performance of NetMHCphosPan over the current state-of-the-art methods PhosMHCpred and NetMHCpan.
In conclusion we have demonstrated the high power of NNAlign_MA for motif deconvolution of complex immuno-peptidomics data, and have developed a novel method for prediction of HLA presented phospho-peptides with improved predictive power and broad HLA coverage. The developed method is available at http://www.cbs.dtu.dk/services/NetMHCphosPan-1.0.
Full text
