CITATIONS
For publication of results, please cite:
-
This version: NetMHCpanExp-1.0. The role of antigen expression in shaping the repertoire of HLA presented ligands.
Heli M. Garcia Alvarez, Zeynep Koşaloğlu-Yalçın, Bjoern Peters, and Morten Nielsen.
In preparation (2022)
DATA RESOURCES
Data resources used to develop this server was obtained from:
- IEDB database.
- Quantitative peptide binding data were obtained
from the IEDB database.
- IMGT/HLA database. Robinson J, Malik A, Parham P, Bodmer JG,
Marsh SGE: IMGT/HLA - a sequence database for the human major histocompatibility complex. Tissue Antigens (2000),
55:280-287.
- HLA protein sequences were obtained from the IMGT/HLA database (version 3.1.0).
- The Human Protein Atlas
Uhlén M et al., Tissue-based map of the human proteome. Science (2015).
- Peptide (or protein) gene expression values are annotated taking as a reference RNA-Seq assays performed over 281 tissue and blood cell samples extracted from the Human Protein Atlas database (v. 20.0).
PORTABLE VERSION
Would you prefer to run NetMHCpanExp at your own site? NetMHCpanExp v. 1.0
is available as a stand-alone software package, with the same
functionality as the service above. Ready-to-ship packages
exist for the most common UNIX platforms. There is a download tap
for academic users; other users are requested to contact
DTU HealthTech Software Package Manager at
health-software@dtu.dk.
INPUT DATA
In this section, the user must define the input for the prediction server following these steps:
1) Specify the desired input data type (FASTA or PEPTIDE) using the drop down menu.
2) Provide the input data by means of pasting the data into the blank field, uploading it using the "Choose File" button or by loading sample data using the "Load Data" button. All the input sequences must be in one-letter amino acid code. The alphabet is as follows (case sensitive):
A C D E F G H I K L M N P Q R S T V W Y and X (unknown)
Any other symbol will be converted to X before processing. At most 5000 sequences are allowed per submission; each sequence must be not more than 20,000 amino acids long and not less than 8 amino acids long.
3) Specify the input format (and peptide length(s) if required):
Sample input data is given after each of the listed options.
a. If FASTA was selected as input type, the user must choose the peptide length(s) the prediction server is going to work with. The input FASTA sequence is digested in overlapping peptides of the provided length(s) and the program predicts binding against all of them. By default input proteins are digested into 9-mer peptides.
Also, the user must decide between these 3 input format options:
- Lookup TPM values per peptide
The digested peptides are searched against the HPA database. Gene expression value annotation for a given peptide is defined as the sum of the TPM values of all protein-coding transcripts containing the exact queried peptide. If a peptide is not found, the program will assign TPM=0.
- Lookup TPM values per protein (using PROT_ID)
The PROT_ID is searched against the HPA database (or the user-specified expression database). Gene expression value annotation is the same for all digested peptides and is equal to the TPM value associated with the queried PROT_ID. If the PROT_ID is not found, the program will assign TPM=0.
Sample input data for options 1. & 2. :
>ENSP00000253039.4
MAGGEAGVTLGQPHLSRQDLTTLDVTKLTPLSHEVISRQATI...
- TPM values given in the input file
The digested peptides are assigned the TPM value annotated in the input file. This number should be a positive rational number. If this is not satisfied, the program will assign TPM=0.
>ENSP00000253039.4 TPM=80.3919983
MAGGEAGVTLGQPHLSRQDLTTLDVTKLTPLSHEVISRQATI...
b. If PEPTIDE was selected as input type, the user must select one of the following 3 input format options:
In all cases, the input format can contain an additional 2nd column that has numerical values. This column can be used to store scores (or target values) that can be futher on employed to compute evaluation metrics.
- [PEP] ([Score])
Peptides are searched against the HPA database. Gene expression value annotation works the same way as described above in a.1.
AAADIVNFL
AAADSIKIW
AAAHFYFEL
AAAPQLLIV
...
- [PEP] ([Score]) [PROT_ID]
PROT_ID(s) are searched against the HPA database (or the user-specified expression database). Gene expression value annotation is performed by assigning to each peptide its corresponding PROT_ID's TPM value. If the PROT_ID is not found, the program will assign TPM=0.
For a list of the Ensembl protein IDs present in the HPA database (genome assembly GRCh38.p12, GENCODE v.28), click HERE.
AAADIVNFL ENSP00000404403.1
AAADSIKIW ENSP00000308179.4
AAAHFYFEL ENSP00000415612.1
AAAPQLLIV ENSP00000252593.6
...
- [PEP] ([Score]) [TPM]
The input is already in the required format for prediction.
AAADIVNFL 0.497551
AAADSIKIW 3.67981
AAAHFYFEL 8.54361
AAAPQLLIV 108.95
...
NOTE: If PEPTIDE was chosen as input type, peptide length selection is unnecessary and thus the corresponding selection box will directly not appear in the interface.

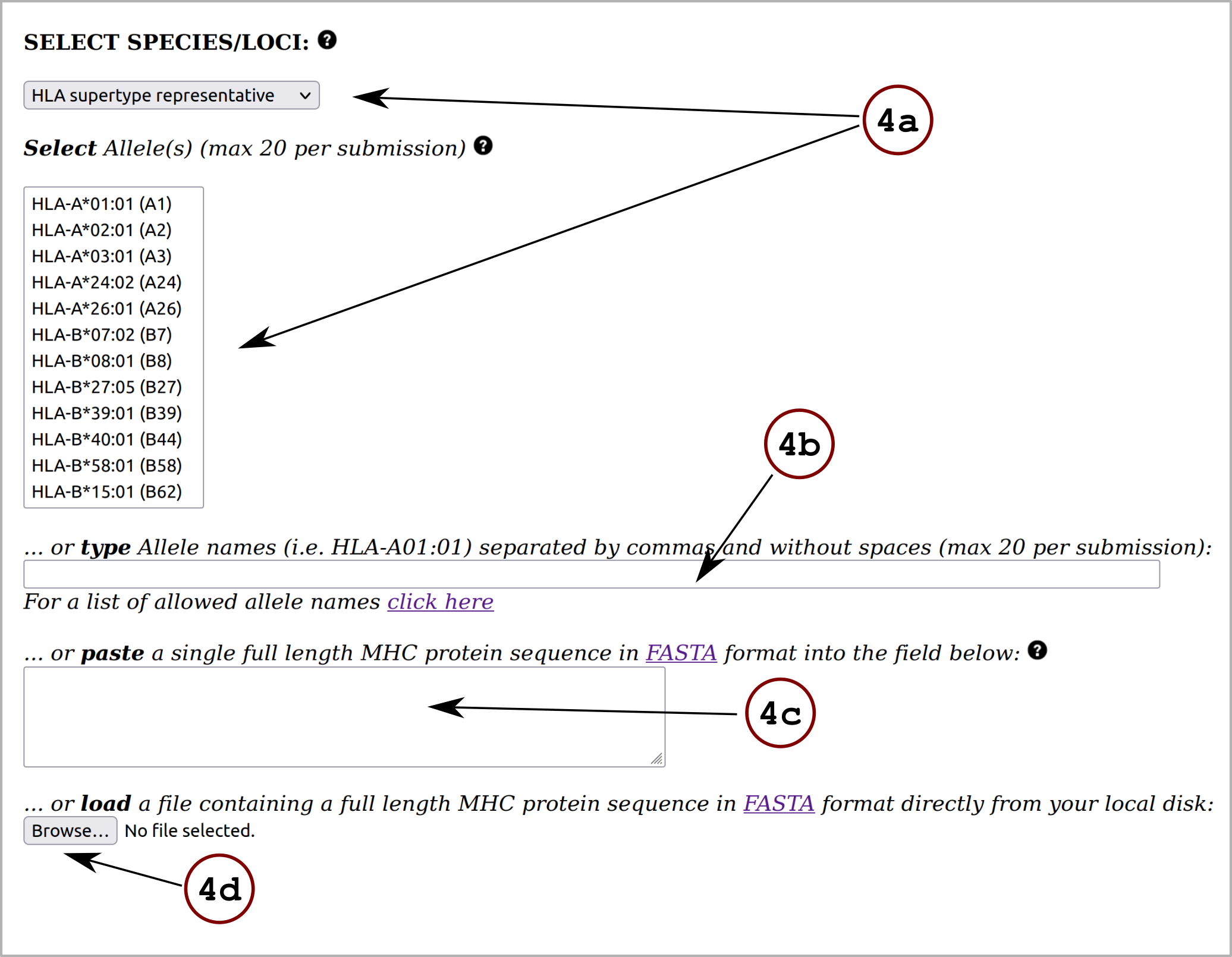
MHC SELECTION
4) The user must define which MHC(s) molecule(s) the input data is going to be predicted against:
a. Select the HLA/MHC supertype family. After choosing the MHC family, you will be able to select a single or multiple MHC molecules from the updated "Select Allele(s)" list.
b. Type the MHC allele names in the provided blank field (separated by commas and without blank spaces). If this is the case, there will be no need to select an MHC supertype familiy from the drop-down menu.
Click here for a list of MHC molecule names. Please note that a maximum of 20 MHC types is allowed per submission.
c. Paste a full MHC protein sequence in the blank box, or
d. directly upload it by clicking the "Choose file" button. Such sequence must be in FASTA format.
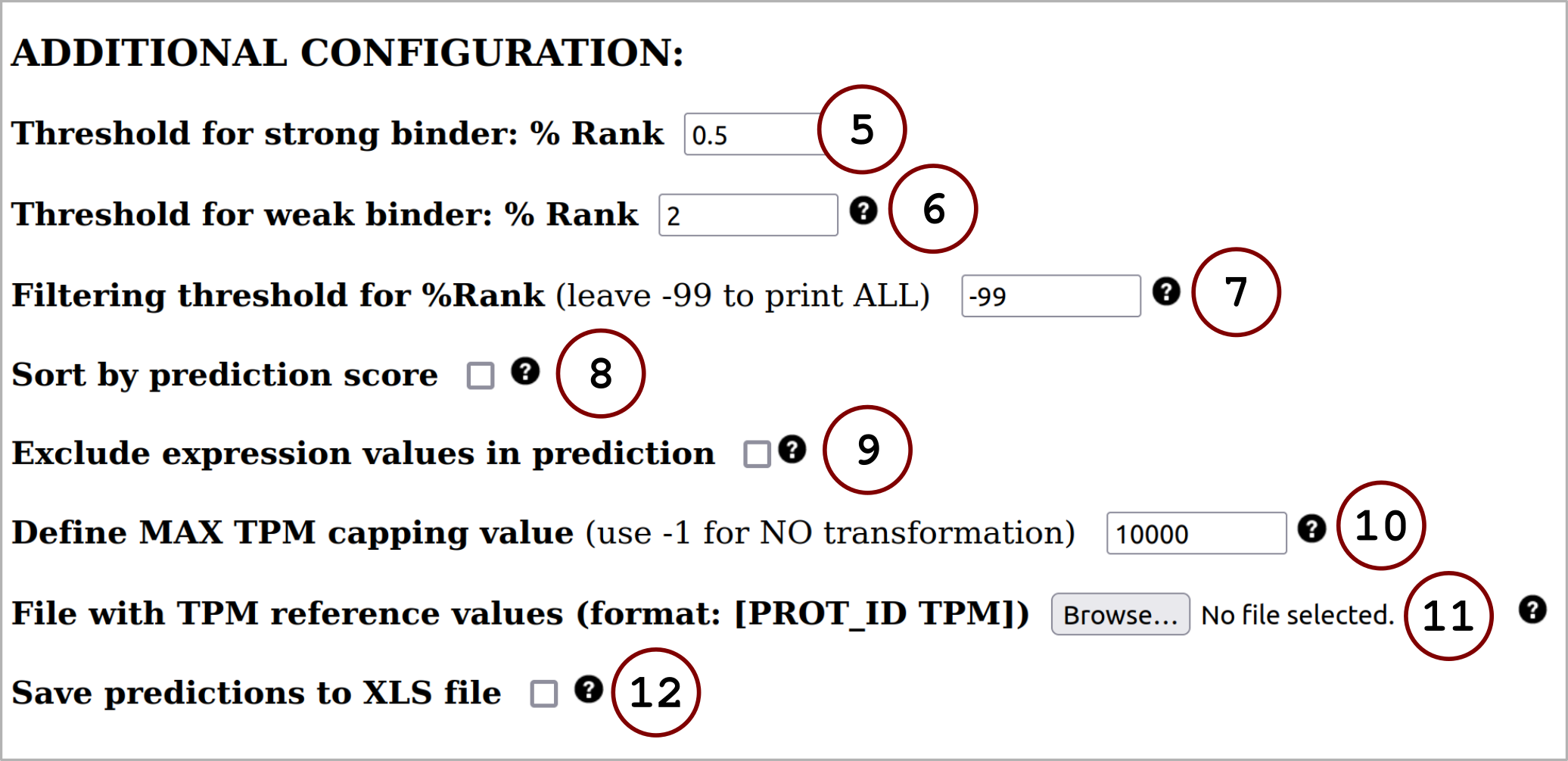
ADDITIONAL CONFIGURATION
In this section, the user may define additional parameters to further customize the run:
5) & 6) Specify thresholds for strong and weak binders. They are expressed in terms of %Rank, that is percentile of the predicted binding affinity compared to the distribution of affinities calculated on set of random natural peptides. The peptide will be identified as a strong binder if it is found among the top x% predicted peptides, where x% is the specified threshold for strong binders (default: 0.5%). The peptide will be identified as a weak binder if the % Rank is above the threshold of the strong binders but below the specified threshold for the weak binders (default: 2%).
7) Specify a %Rank threshold to filter out predictions. Only sequences with a predicted %Rank value less than the specified threshold will be printed. To print all predictions, leave this value set to -99.
8) Tick this box to have the output sorted by descending prediction score.
9) Tick this box to exclude expression values from the prediction. The method will ignore expression values in the input file, if present.
10) Specify the maximum TPM value to map input TPM values to the [0,1] interval. If the values already fall within this unit range, set this option to -1.
11) Load a file with a reference protein expression database. The file must contain two columns: [PROT_ID] [TPM]. This file will be used perform automatic TPM value annotation of input peptides or proteins (FASTA format). Be aware that the input peptides (or proteins) must have PROT_ID(s) that match the ones present in the provided database.
12) Enable this option to export the prediction output to .XLS format (readable by most spreadsheet softwares, like Microsoft Excel).
SUBMISSION
After the user has finished the "INPUT DATA", "MHC SELECTION" and "ADDITIONAL CONFIGURATION" steps, the submission can now be done. To do so, the user can click on "Submit" to submit the job to the processing server, or click on "Clear fields" to clear the page and start over.
The status of your job (either 'queued' or 'running') will be displayed and constantly updated until it terminates and the server output appears in the browser window.
After the server has finished running the corresponding predictions, an output page will be delivered to the user. A description of the output format can be found at output format HERE.
At any time during the wait you may enter your e-mail address and simply leave the window. Your job will continue; when it terminates you will be notified by e-mail with a URL to your results. They will be stored on the server for 24 hours.
OUTPUT EXAMPLE
For the following
FASTA input example:
>ENSP00000253039.4 TPM=80.3919983
MAGGEAGVTLGQPHLSRQDLTTLDVTKLTPLSHEVISRQATINIGTIGHVAHGKSTVVKAISGVHTVRFKNELERNITIKLGYANAKIYKLDDPSCPRPE
With parameters:
- Peptide length: 8, 9, 10, 11
- Input format: TPM values given in the input file
- Allele: HLA-A*0201
- Threshold for strong binder: 0.5 %Rank
- Threshold for weak binder: 2.0 %Rank
- Filtering threshold for %Rank: -99
- Sort by prediction score: On
- Exclude Expression in prediction: Off
- Define MAX TPM capping value: 10000
NetMHCpanExp-1.0 will return the following output (showing the first 15 predicted peptides):
# NetMHCpanExp version 1.0
# Tmpdir made /var/www/webface/tmp/server/netmhcpanexp/6237B40000005ED4FDF6B89C/netMHCpanv9J7VL
# Input is in FSA format
# Peptide length 8,9,10,11
# Make predictions including expression
HLA-A02:01 : Distance to training data 0.000 (using nearest neighbor HLA-A02:01)
# Rank Threshold for Strong binding peptides 0.500
# Rank Threshold for Weak binding peptides 2.000
------------------------------------------------------------------------------------------------------------
Pos HLA Peptide Core Of Gp Gl Ip Il Icore Identity Score_EL Rnk_EL Exp BindLevel
------------------------------------------------------------------------------------------------------------
27 HLA-A*02:01 KLTPLSHEV KLTPLSHEV 0 0 0 0 0 KLTPLSHEV ENSP00000253039 0.9951022 0.009 0.47764 <= SB
59 HLA-A*02:01 KAISGVHTV KAISGVHTV 0 0 0 0 0 KAISGVHTV ENSP00000253039 0.9754907 0.074 0.47764 <= SB
83 HLA-A*02:01 YANAKIYKL YANAKIYKL 0 0 0 0 0 YANAKIYKL ENSP00000253039 0.9058493 0.237 0.47764 <= SB
56 HLA-A*02:01 TVVKAISGV TVVKAISGV 0 0 0 0 0 TVVKAISGV ENSP00000253039 0.7207928 0.635 0.47764 <= WB
57 HLA-A*02:01 VVKAISGVHTV VVISGVHTV 0 2 2 0 0 VVKAISGVHTV ENSP00000253039 0.6767913 0.746 0.47764 <= WB
25 HLA-A*02:01 VTKLTPLSHEV VLTPLSHEV 0 1 2 0 0 VTKLTPLSHEV ENSP00000253039 0.6566866 0.793 0.47764 <= WB
26 HLA-A*02:01 TKLTPLSHEV TLTPLSHEV 0 1 1 0 0 TKLTPLSHEV ENSP00000253039 0.5708980 0.988 0.47764 <= WB
27 HLA-A*02:01 KLTPLSHEVI KLTPLSHEI 0 8 1 0 0 KLTPLSHEVI ENSP00000253039 0.5401132 1.077 0.47764 <= WB
80 HLA-A*02:01 KLGYANAKI KLGYANAKI 0 0 0 0 0 KLGYANAKI ENSP00000253039 0.4893585 1.206 0.47764 <= WB
14 HLA-A*02:01 HLSRQDLTTL HLSQDLTTL 0 3 1 0 0 HLSRQDLTTL ENSP00000253039 0.4232187 1.430 0.47764 <= WB
60 HLA-A*02:01 AISGVHTV AI-SGVHTV 0 0 0 2 1 AISGVHTV ENSP00000253039 0.3835837 1.575 0.47764 <= WB
22 HLA-A*02:01 TLDVTKLTPL TLDVTKLTL 0 8 1 0 0 TLDVTKLTPL ENSP00000253039 0.3285049 1.809 0.47764 <= WB
58 HLA-A*02:01 VKAISGVHTV VAISGVHTV 0 1 1 0 0 VKAISGVHTV ENSP00000253039 0.2115432 2.467 0.47764
55 HLA-A*02:01 STVVKAISGV SVVKAISGV 0 1 1 0 0 STVVKAISGV ENSP00000253039 0.1888403 2.654 0.47764
40 HLA-A*02:01 ATINIGTIGHV ATINIGTHV 0 7 2 0 0 ATINIGTIGHV ENSP00000253039 0.1826325 2.710 0.47764
DESCRIPTION
The prediction output consists of the following columns:
Pos: Residue number (starting from 0) of the peptide in the protein sequence (if the input is in peptide format, dismiss this column).
HLA: Specified MHC molecule / Allele name.
Peptide: Amino acid sequence of the potential ligand.
Core: The minimal 9 amino acid binding core directly in contact with the MHC.
Of: The starting position of the Core within the Peptide (if > 0, the method predicts a N-terminal protrusion).
Gp: Position of the deletion, if any.
Gl: Length of the deletion, if any.
Ip: Position of the insertion, if any.
Il: Length of the insertion, if any.
Icore: Interaction core. This is the sequence of the binding core including eventual insertions of deletions.
Identity: Protein identifier, i.e. the name of the FASTA entry.
Score: The raw prediction score.
%Rank: Rank of the predicted binding score compared to a set of random natural peptides for the corresponding MHC molecule. This measure is not affected by the inherent bias of certain molecules towards higher or lower mean predicted scores. We advise to select candidate binders based on %Rank rather than Score.
Exp: The gene expression value in TPM normalized by the maximum TPM value specified by the user (default: 10000 TPM). If the expression values were excluded from the prediction, this column will not appear in the output.
BindLevel: (SB: Strong Binder, WB: Weak Binder). The peptide will be identified as a strong binder if the %Rank is below the specified threshold for the strong binders (default: 0.5%). The peptide will be identified as a weak binder if the %Rank is above the threshold of the strong binders but below the specified threshold for the weak binders (default: 2%).
NOTES
Peptide vs. iCore vs. Core
Three amino acid sequences are reported for each peptide (row) in the prediction output:
- The Peptide is the complete amino acid sequence evaluated by NetMHCpanExp. Peptides are the
full sequences submitted as a peptide list, or the result of the digestion of source proteins (submission in FASTA format)
- The iCore is a substring of Peptide, encompassing
all residues between P1 and P-omega interacting with the MHC. For all intents and purposes, this is the minimal candidate
ligand that should be considered for further validation.
- The Core is always 9 amino acids long and is a construction used for sequence alignment and identification of binding anchors.
ARTICLE ABSTRACTS
MAIN REFERENCE
NetMHCpanExp-1.0. The role of antigen expression in shaping the repertoire of HLA presented ligands.
Heli M. Garcia Alvarez 1, Zeynep Koşaloğlu-Yalçın 2, Bjoern Peters2,3, and Morten Nielsen1,4
1
Instituto de Investigaciones Biotecnológicas, Universidad Nacional de San Martín, Buenos Aires, BA 16503, Argentina.
2
Center for Infectious Disease and Vaccine Research, La Jolla Institute for Immunology, La Jolla, California, USA
3
Department of Medicine, University of California, San Diego, La Jolla, California, USA
4
Department of Bio and Health Informatics, Technical University of Denmark, Kgs. Lyngby, DK 28002, Denmark.
In preparation (2022)
OTHER RELEVANT REFERENCES
NNAlign_MA; MHC Peptidome Deconvolution for Accurate MHC Binding Motif Characterization and Improved T-cell Epitope Predictions.
Bruno Alvarez 1, Birkir Reynisson 2, Carolina Barra 1, Søren Buus 3, Nicola Ternette 4, Tim Connelley 5, Massimo Andreatta 1, Morten Nielsen 1,2
1
Instituto de Investigaciones Biotecnológicas, Universidad Nacional de San Martín, Buenos Aires, BA 16503, Argentina.
2
Department of Bio and Health Informatics, Technical University of Denmark, Lyngby, Denmark.
3
Department of Immunology and Microbiology, Faculty of Health Sciences, University of Copenhagen, Denmark.
4
The Jenner Institute, Nuffield Department of Medicine, Oxford, United Kingdom.
5
Roslin Institute, Edinburgh, Midlothian, United Kingdom.
Mol Cell Proteomics (2019); 8(12):2459-2477. DOI: 10.1074/mcp.TIR119.001658. PMID: 31578220
The set of peptides presented on a cell's surface by MHC molecules is known as the immunopeptidome. Current mass spectrometry technologies allow for identification of large peptidomes, and studies have proven these data to be a rich source of information for learning the rules of MHC-mediated antigen presentation. Immunopeptidomes are usually poly-specific, containing multiple sequence motifs matching the MHC molecules expressed in the system under investigation. Motif deconvolution -the process of associating each ligand to its presenting MHC molecule(s)- is therefore a critical and challenging step in the analysis of MS-eluted MHC ligand data. Here, we describe NNAlign_MA, a computational method designed to address this challenge and fully benefit from large, poly-specific data sets of MS-eluted ligands. NNAlign_MA simultaneously performs the tasks of (1) clustering peptides into individual specificities; (2) automatic annotation of each cluster to an MHC molecule; and (3) training of a prediction model covering all MHCs present in the training set. NNAlign_MA was benchmarked on large and diverse data sets, covering class I and class II data. In all cases, the method was demonstrated to outperform state-of-the-art methods, effectively expanding the coverage of alleles for which accurate predictions can be made, resulting in improved identification of both eluted ligands and T-cell epitopes. Given its high flexibility and ease of use, we expect NNAlign_MA to serve as an effective tool to increase our understanding of the rules of MHC antigen presentation and guide the development of novel T-cell-based therapeutics.
Supplementary material
Here you will find the datasets used for the independent evaluation of NetMHCpanExp-1.0 method.
MS Ligands
Test_set_I-B_(HPA)
Test_set_I-B_(INT)
Test_set_I-B_(PAXdb)
Test_set_I-C_CLL_(HPA)
Test_set_I-C_CLL_(INT)
Test_set_I-C_CLL_(PAXdb)
Test_set_I-C_OV_(HPA)
Test_set_I-C_OV_(INT)
Test_set_I-C_OV_(PAXdb)
(Format: [PEPTIDE] [TARGET_VALUE] [TEST_SET_No] [GENE EXP (TPM)])
Test_set_I-NCI
(Format: [PEPTIDE] [TARGET_VALUE] [MHC_ALLELE] [GENE EXP (TPM)])
CD8+ Epitopes
CD8+_epitopes_IEDB
CD8+_epitopes_YFV
(Format: [PEPTIDE] [TARGET_VALUE] [MHC_ALLELE])
Download ALL evaluation data:
Supplementary_Data
Submitted (2022)