INPUT DATA
In this section, the user must define the input for the prediction server following these steps:
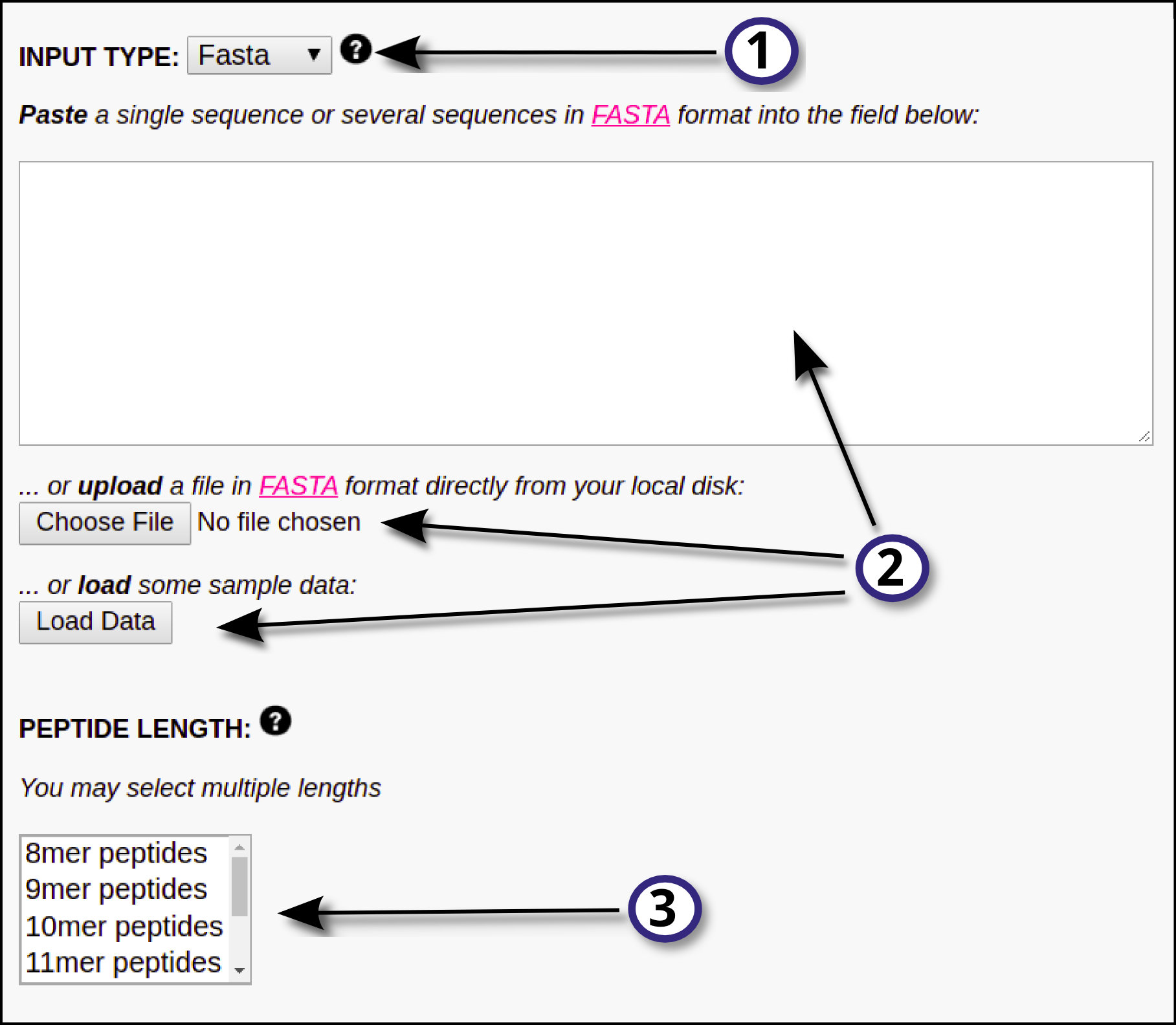
1) Specify the desired type of input data (FASTA or PEPTIDE) using the drop down menu.
2) Provide the input data by means of pasting the data into the blank field, uploading it using the "Choose File" button or by loading sample data using the "Load Data" button. All the input sequences must be in one-letter amino acid code. The alphabet is as follows (case sensitive):
A C D E F G H I K L M N P Q R S T V W Y and X (unknown)
Any other symbol will be converted to X before processing. At most 5000 sequences are allowed per submission; each sequence must be not more than 20,000 amino acids long and not less than 8 amino acids long.
3) If FASTA was selected as input type, the user must select the peptide length(s) the prediction server is going to work with. NetMHCpan-4.1 will "chop" the input FASTA sequence in overlapping peptides of the provided length(s) and will predict binding against all of them. By default input proteins are digested into 9-mer peptides. Note that, if PEPTIDE was selected as input type, this step is unnecessary and thus the peptide length selector will directly not appear in the interface.
MHC SELECTION
Here, the user must define which MHC(s) molecule(s) the input data is going to be predicted against:
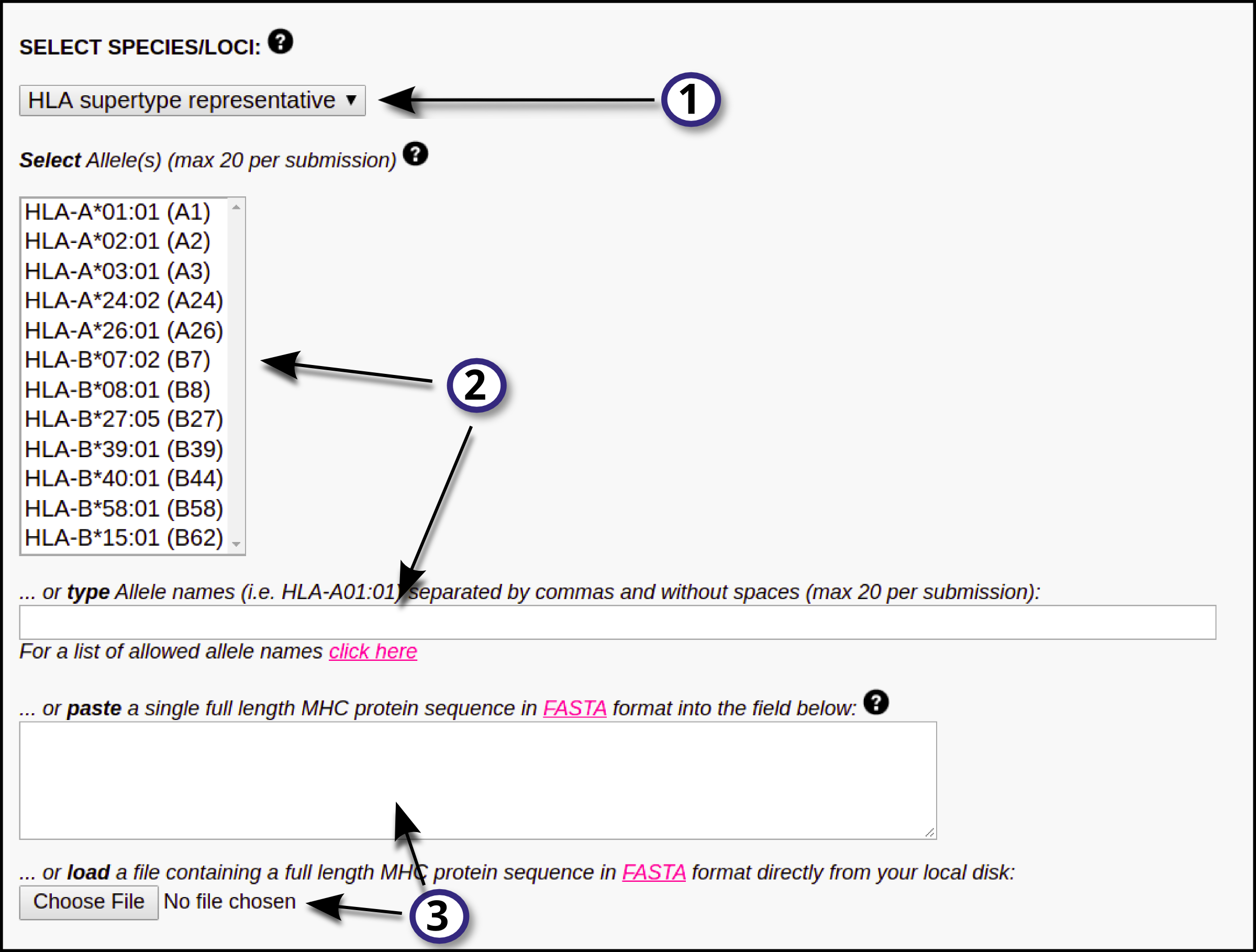
1) First, select the HLA/MHC supertype family.
2) After selecting the MHC family, the user will be able to select a single or multiple MHC molecules from the updated "Select Allele(s)" list. On the other hand, the user may opt to directly type the MHC names in the provided blank field (separated by commas and without blank spaces); if this is the case, there will be no need to select an MHC supertype familiy from the drop-down menu. Click here for a list of MHC molecule names (use the names in the first column). Please note that a maximum of 20 MHC types is allowed per submission.
3) Optionally, the user may choose to paste a full MHC protein sequence in the blank box, or directly upload it by clicking the "Choose file" button. Such sequence must be in FASTA format.
Please note that steps 2) and 3) are mutually exclusive, and are only labeled this way for explanation purposes.
ADDITIONAL CONFIGURATION
In this section, the user may define additional parameters to further customize the run:
1, 2) Specify thresholds for strong and weak binders. They are expressed in terms of %Rank, that is percentile of the predicted binding affinity compared to the distribution of affinities calculated on set of random natural peptides. The peptide will be identified as a strong binder if it is found among the top x% predicted peptides, where x% is the specified threshold for strong binders (by default 0.5%). The peptide will be identified as a weak binder if the % Rank is above the threshold of the strong binders but below the specified threshold for the weak binders (by default 2%).
3) Specify a %Rank threshold to filter out predictions. Only sequences with a predicted %Rank value less than the specified threshold will be printed. To print all predictions, leave this value set to -99.
4) Tick this option to include also Binding Affinity predictions together with Eluted Ligand likelihood.
5) Tick this box to have the output sorted by descending prediction score.
6) Enable this option to export the prediction output to .XLS format (readable by most spreadsheet softwares, like Microsoft Excel).

SUBMISSION
After the user has finished the "INPUT DATA", "MHC SELECTION" and "ADDITIONAL CONFIGURATION" steps, the submission can now be done. To do so, the user can click on "Submit" to submit the job to the processing server, or click on "Clear fields" to clear the page and start over.
The status of your job (either 'queued' or 'running') will be displayed and constantly updated until it terminates and the server output appears in the browser window.
After the server has finished running the corresponding predictions, an
output page will be delivered to the user. A description of the output format can be found at output format
At any time during the wait you may enter your e-mail address and simply leave the window. Your job will continue; when it terminates you will be notified by e-mail with a URL to your results. They will be stored on the server for 24 hours.
