CITATIONS
For publication of results, please cite:
-
Improved prediction of MHC II antigen presentation through integration and motif deconvolution of mass spectrometry MHC eluted ligand data.
Reynisson B, Barra C, Kaabinejadian S, Hildebrand WH, Peters B, Nielsen M
J Proteome Res 2020 Apr 30. doi: 10.1021/acs.jproteome.9b00874.
PubMed: 32308001
DATA RESOURCES
Benchmark data used to develop this server were obtained from:
PORTABLE VERSION
Output format
EXAMPLE OUTPUT
For the following FASTA input example:
>P9WNK5
MAEMKTDAATLAQEAGNFERISGDLKTQIDQVESTAGSLQGQWRGAAGTAAQAAVVRFQEAANKQKQELDEISTNIRQAGVQYSRADEEQQQALSSQMGF
With parameters:
Peptide length: 15
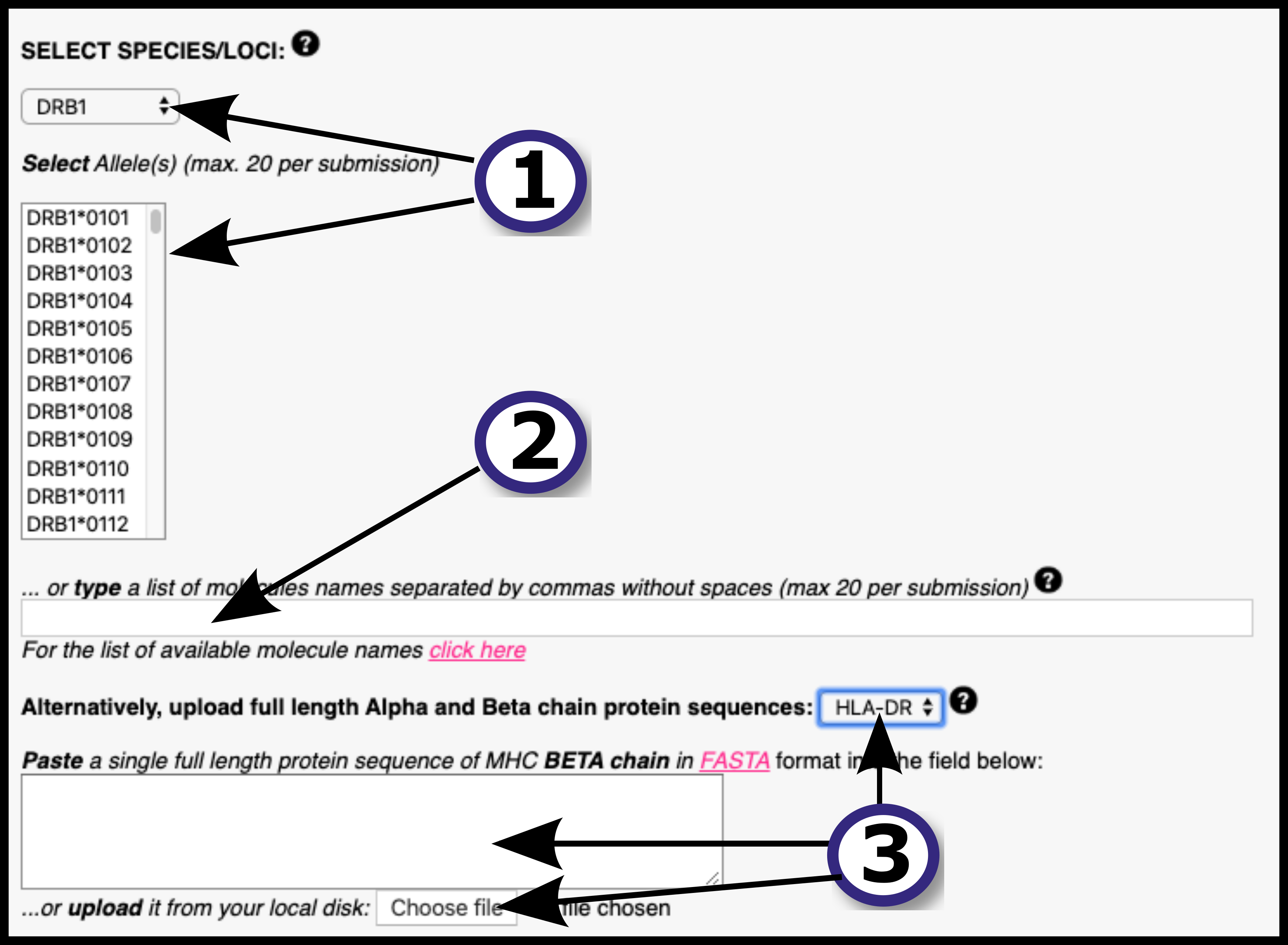
Allele: DRB1_0101
Sort by prediction score: On
NetMHCIIpan-4.1 will return the following output (showing the top 10 predicted peptides):
# NetMHCIIpan version 4.1
# Input is in FASTA format
# Peptide length 15
# Prediction Mode: EL
# Threshold for Strong binding peptides (%Rank) 1%
# Threshold for Weak binding peptides (%Rank) 5%
# Allele: DRB1_0101
--------------------------------------------------------------------------------------------------------------------------------------------
Pos MHC Peptide Of Core Core_Rel Identity Score_EL %Rank_EL Exp_Bind BindLevel
--------------------------------------------------------------------------------------------------------------------------------------------
40 DRB1_0101 QGQWRGAAGTAAQAA 3 WRGAAGTAA 1.000 P9WNK5 0.867119 0.56 NA <=SB
39 DRB1_0101 LQGQWRGAAGTAAQA 4 WRGAAGTAA 1.000 P9WNK5 0.775118 0.99 NA <=SB
41 DRB1_0101 GQWRGAAGTAAQAAV 2 WRGAAGTAA 1.000 P9WNK5 0.585897 2.04 NA <=WB
38 DRB1_0101 SLQGQWRGAAGTAAQ 5 WRGAAGTAA 1.000 P9WNK5 0.513786 2.57 NA <=WB
73 DRB1_0101 STNIRQAGVQYSRAD 3 IRQAGVQYS 1.000 P9WNK5 0.184990 7.08 NA
26 DRB1_0101 KTQIDQVESTAGSLQ 3 IDQVESTAG 0.993 P9WNK5 0.122282 9.40 NA
15 DRB1_0101 AGNFERISGDLKTQI 3 FERISGDLK 1.000 P9WNK5 0.121026 9.45 NA
53 DRB1_0101 AAVVRFQEAANKQKQ 3 VRFQEAANK 0.873 P9WNK5 0.118252 9.59 NA
14 DRB1_0101 EAGNFERISGDLKTQ 4 FERISGDLK 1.000 P9WNK5 0.118084 9.60 NA
72 DRB1_0101 ISTNIRQAGVQYSRA 4 IRQAGVQYS 0.993 P9WNK5 0.103021 10.47 NA
DESCRIPTION
The prediction output for each molecule consists of the following columns:
Pos Residue number (starting from 0)
MHC MHC molecule name
Peptide Amino acid sequence
Of Starting position offset of the optimal binding core (starting from 0)
Core Binding core register
Core_Rel Reliability of the binding core, expressed as the fraction of networks in the ensemble selecting the optimal core
Identity Annotation of the input sequence, if specified
Score_EL Eluted ligand prediction score
%Rank_EL Percentile rank of eluted ligand prediction score
Exp_bind If the input was given in PEPTIDE format with an annotated affinity value (mainly for benchmarking purposes).
Score_BA Predicted binding affinity in log-scale (printed only if binding affinity predictions were selected)
Affinity(nM) Predicted binding affinity in nanomolar IC50 (printed only if binding affinity predictions were selected)
%Rank_BA % Rank of predicted affinity compared to a set of 100.000 random natural peptides. This measure is not affected by inherent bias of certain molecules towards higher or lower mean predicted affinities (printed only if binding affinity predictions were selected)
BindLevel (SB: strong binder, WB: weak binder). The peptide will be identified as a strong binder if the % Rank is below the specified threshold for the strong binders. The peptide will be identified as a weak binder if the % Rank is above the threshold of the strong binders but below the specified threshold for the weak binders.
Supplementary material
Training data
NetMHCIIpan-4.1
Here, you will find the data set used for training of NetMHCIIpan-4.1.
NetMHCIIpan_train.tar.gz
Download the file and untar the content using
cat NetMHCIIpan_train.tar.gz | uncompress | tar xvf -
This will creat the directory called NetMHCIIpan_train. In this directory you will find 12 files. 10 files (c00?_ba, c00?_el) with partitions with binding affinity (ba) with eluted ligand data (el). The format for each file is (here shown for an el file)
AAAAAAAAAAAAA 1 Bergseng__9037_SWEIG AGRAAAAAAAAG
AAAAAAAAAAAAA 1 Bergseng__9064_AMALA AGRAAAAAAAAG
AAAAAAAAAAAAA 1 Bergseng__9089_BOB AGRAAAAAAAAG
AAAAAAAAAAAAAA 1 Bergseng__9037_SWEIG AGRAAAAAAAGA
AAAAAAAAAAAAAA 1 Bergseng__9064_AMALA AGRAAAAAAAGA
AAAAAAAAAAAAAA 1 Bergseng__9089_BOB AGRAAAAAAAGA
AAAAAAAAAAAAAAA 1 Bergseng__9037_SWEIG AGRAAAAAAGAG
AAAAAAAAAAAAAAA 1 Bergseng__9064_AMALA AGRAAAAAAGAG
AAAAAAAAAAAAAAA 1 Bergseng__9089_BOB AGRAAAAAAGAG
AAAAAAAAAAAAAAAAAAAAA 1 Abelin__MAPTAC_HLA_DQB10602_DQA10102 KHPAAAAAAYYQ
where the different columns are peptide, target value, MHC_molecule/cell-line, and
context. In cases where the 3rd columns is a cell-line ID, the MHC molecules
expressed in the cell-line are listed in the allelelist.txt file.
The allelelist.txt file contains the information about alleles expressed in each MA
cell-line data set, and pseudosequence.2016.all.X.dat the MHC pseudo sequenes for each
MHC molecule.
Version history
Please click on the version number to activate the corresponding server.
|
4.0
|
The current server (online since April 2020). New in this version:
- The two output neuron architechture introduced in NetMHCpan-4.0 permits the inclusion of EL data, and the new training algorithm NNAlign_MA extends training data to ligands of ambiguous allele assignments. The model also, optionally, encodes ligand context.
Main publication:
-
Improved prediction of MHC II antigen presentation through integration and motif deconvolution of mass spectrometry MHC eluted ligand data.
Reynisson B, Barra C, Kaabinejadian S, Hildebrand WH, Peters B, Nielsen M
J Proteome Res 2020 Apr 30. doi: 10.1021/acs.jproteome.9b00874.
PubMed: 32308001
|
|
3.2
|
(online since January 2018). New in this version:
- Method retrained on an extensive dataset of over 100,000 datapoints, covering 36 HLA-DR, 27 HLA-DQ, 9 HLA-DP, and 8 mouse MHC-II molecules.
Main publication:
-
Improved methods for predicting peptide binding affinity to MHC class II molecules.
Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, Sette A, Peters B, Nielsen M.
Immunology. 2018 Jan 6. doi: 10.1111/imm.12889.
PubMed: 29315598
|
|
3.1
|
(online since December 2014). New in this version:
- Improved binding core identification by realigning individual networks in the ensemble.
- Introduced a reliability measure on the predicted binding core (Core_Rel column).
- Graphical representation of the binding core register and of possible multiple cores.
Main publication:
-
Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification
Andreatta M, Karosiene E, Rasmussen M, Stryhn A, Buus S, and Nielsen M
Immunogenetics (2015)
PubMed: 26416257
|
|
3.0
|
(online since June 2013). New in this version:
- The user can make predictions for all DR, DP and DQ molecules with known protein
sequence. Likewise can the user upload full length MHC class II alpha and beta chain and have the server predict MHC restricted peptides from any given protein of interest
|
|
2.1
|
(online since 6 June 2011). New in this version:
- User can upload full length MHC class II beta chain and have the server predict MHC restricted peptides from any given protein of interest.
|
|
2.0
|
(online since 17 Nov 2010). New in this version:
- New concurent algorithm used to train the network.
|
|
1.1
|
(online since 15 April 2010). New in this version:
- %-rank measure include for each prediction value. The %-rank score give the rank of the
prediction score to a distribution of prediction scores from 200.000 natural random 15mer peptides.
|
|
1.0
|
Original version (online version until April 15 2010):
Main publication:
-
Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan.
Nielsen M, et al. (2008) PLoS Comput Biol. Jul 4;4(7):e1000107.
View the abstract, the full text version at PLoS Compu:
Full text.
|