INPUT DATA
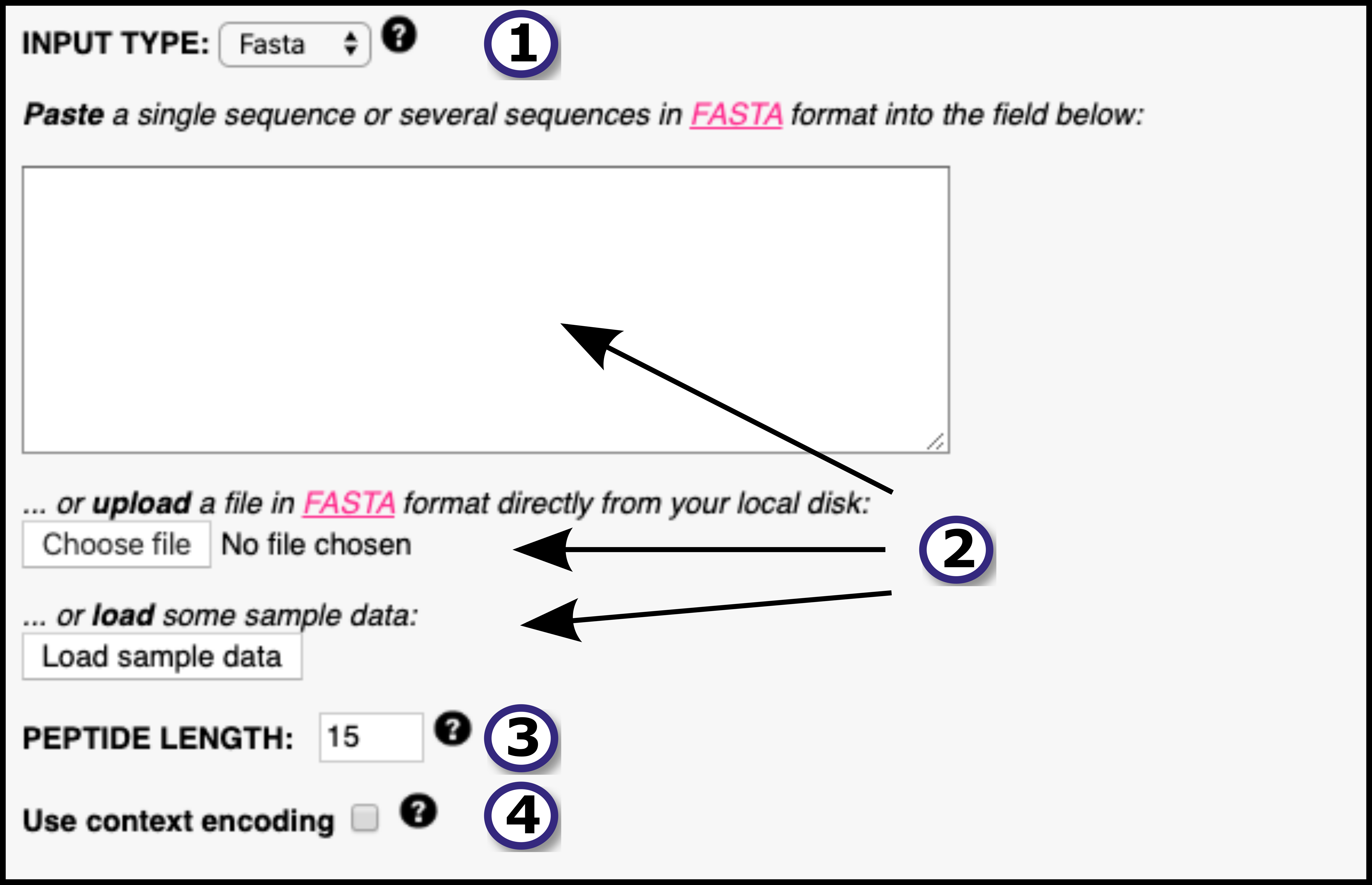
In this section, the user must define the input for the prediction server following these steps:1) Specify the desired type of input data (FASTA or PEPTIDE ) using the drop down menu.
2) Provide the input data by means of pasting the data into the blank field, uploading it using the "Choose File" button or by loading sample data using the "Load Data" button. All the input sequences must be in one-letter amino acid code. The alphabet is as follows (case sensitive):
A C D E F G H I K L M N P Q R S T V W Y and X (unknown)
Any other symbol will be converted to X before processing. At most 5000 sequences are allowed per submission; each sequence must be not more than 20,000 amino acids long and not less than 9 amino acids long.
3) If FASTA was selected as input type, the user must select the peptide length(s) the prediction server is going to work with. NetMHCIIpan-4.0 will "chop" the input FASTA sequence in overlapping peptides of the provided length and will predict binding against all of them. By default input proteins are digested into 15-mer peptides. Note that, if PEPTIDE was selected as input type, this step is unnecessary and thus the peptide length selector will directly not appear in the interface.
4)Context encoding informs the network of the proteolytic context the ligand. Context is automatically generated from the source protein if the user selects FASTA format. Briefly, context is made up of 12 amino acids: 3 amino acids upstream of the ligand, 3 first amino acids at the ligand N-terminus, 3 last amino acids at the ligand C-terminus and 3 amino acids downstream the ligand(in the source protein), all concatenated together. If the input type is PEPTIDE , the user must specify the ligand context(see PEPTIDECONT ).