Submission
CITATIONS AND FUNDING
Developed under the following contracts:
* National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, contract No. HHSN272201200010C
* Agencia Nacional de Promoción Científica y Tecnológica, Argentina (PICT-2012-0115)
For publication of results, please cite:
-
Gapped sequence alignment using artificial neural networks: application to the MHC class I system.
Andreatta M, Nielsen M
Bioinformatics (2016) Feb 15;32(4):511-7
PubMed:
26515819
[PDF]
-
Reliable prediction of T-cell epitopes using neural networks with novel
sequence representations.
Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S,
Brunak S, Lund O.
Protein Sci., (2003) 12:1007-17
PubMed:
12717023
Instructions
1. Specify the input sequences
All the input sequences must be in one-letter amino acid
code. The alphabet is as follows
(case sensitive):
A C D E F G H I K L M N P Q R S T V W Y and X (unknown)
Any other symbol will be converted to X before processing.
The server allows for input in either FASTA or
PEPTIDE format.
Sequences can be submitted in the following two formats:
-
Paste a single sequence (just the amino acids) or a number of sequences in
FASTA
format or a list of peptides into the upper window of the main server page.
-
Select a FASTA
or PEPTIDE
file on your local disk, either by typing the file name into the lower window
or by browsing the disk.
At most 5000 sequences per submission; each sequence not more than 20,000 amino acids and not less than 8 amino acids.
2. Customize your run
1. Specify peptide length (only for FASTA input). By default input proteins are digested into 9-mer peptides.
2. Select species/loci from the scroll-down menu.
3. Select allele(s) from the scroll-down menu or type in the allele names separated by commas (without blank spaces). If you choose to type in the allele names, you can consult the List of MHC molecule names.; use the molecule names in the first column.
4. Optionally specify thresholds for strong and weak binders. They are expressed in terms of %Rank, that is percentile of the predicted binding affinity compared to the distribution of affinities calculated on set of 400.000 random natural peptides. The peptide will be identified as a strong binder if it is found among the top x% predicted peptides, where x% is the specified threshold for strong binders (by default 0.5%). The peptide will be identified as a weak binder if the % Rank is above the threshold of the strong binders but below the specified threshold for the weak binders (by default 2%).
5. Tick the box Sort by affinity to have the output sorted by descending predicted binding affinity.
3. Submit the job
Click on the
"Submit" button. The status of your job (either 'queued'
or 'running') will be displayed and constantly updated until it terminates and
the server output appears in the browser window.
At any time during the wait you may enter your e-mail address and simply leave
the window. Your job will continue; when it terminates you will be notified by e-mail with a URL to your results. They will be stored on the server for 24 hours.
4. Output
A description of the output format can be found in the Output format tab.
Format of NetMHC-4.0 output
DESCRIPTION
The prediction output for each molecule consists of the following columns:
Pos Residue number (starting from 0)
HLA Molecule/allele name
Peptide Amino acid sequence of the potential ligand
Core The minimal 9 amino acid binding core directly in contact with the MHC
Offset The starting position of the Core within the Peptide (if > 0, the method predicts a N-terminal protrusion)
I_pos Position of the insertion, if any.
I_len Length of the insertion.
D_pos Position of the deletion, if any.
D_len Length of the deletion.
iCore Interaction core. This is the sequence of the binding core including eventual insertions of deletions.
Identity Protein identifier, i.e. the name of the Fasta entry.
1-log50k(aff) Log-transformed binding affinity. Some reference transformations: 50,000nM -> logAff=0; 500nM -> logAff=0.426; 50nM -> logAff=0.638; 1nM -> logAff=1.000.
Affinity(nM) Predicted binding affinity in nanoMolar units.
%Rank Rank of the predicted affinity compared to a set of 400.000 random natural peptides. This measure is not affected by inherent bias of certain molecules towards higher or lower mean predicted affinities. Strong binders are defined as having %rank<0.5, and weak binders with %rank<2. We advise to select candidate binders based on %Rank rather than nM Affinity
BindLevel (SB: strong binder, WB: weak binder). The peptide will be identified as a strong binder if the % Rank is below the specified threshold for the strong binders, by default 0.5%. The peptide will be identified as a weak binder if the % Rank is above the threshold of the strong binders but below the specified threshold for the weak binders, by default 2%.
EXAMPLE OUTPUT
Fasta input:
>Gag_180_209
TPQDLNTMLNTVGGHQAAMQMLKETINEEA
Peptide length:
8 and
9
Allele:
HLA-A*0301
will return the following predictions:
# NetMHC version 4.0
# Input is in FSA format
# Peptide length 8,9
# Affinity Threshold for Strong binding peptides 50.000
# Affinity Threshold for Weak binding peptides 500.000
# Rank Threshold for Strong binding peptides 0.500
# Rank Threshold for Weak binding peptides 2.000
-----------------------------------------------------------------------------------
pos HLA peptide Core Offset I_pos I_len D_pos D_len iCore Identity 1-log50k(aff) Affinity(nM) %Rank BindLevel
-----------------------------------------------------------------------------------
0 HLA-A0301 TPQDLNTM -TPQDLNTM 0 0 1 0 0 TPQDLNTM Gag_180_209 0.014 43017.00 95.00
1 HLA-A0301 PQDLNTML PQDLNTML- 0 8 1 0 0 PQDLNTML Gag_180_209 0.021 39881.02 80.00
2 HLA-A0301 QDLNTMLN -QDLNTMLN 0 0 1 0 0 QDLNTMLN Gag_180_209 0.018 41073.47 85.00
3 HLA-A0301 DLNTMLNT DLN-TMLNT 0 3 1 0 0 DLNTMLNT Gag_180_209 0.019 40552.86 85.00
4 HLA-A0301 LNTMLNTV -LNTMLNTV 0 0 1 0 0 LNTMLNTV Gag_180_209 0.035 34098.43 55.00
5 HLA-A0301 NTMLNTVG NTMLNTVG- 0 8 1 0 0 NTMLNTVG Gag_180_209 0.025 38038.41 70.00
6 HLA-A0301 TMLNTVGG TMLNTVGG- 0 8 1 0 0 TMLNTVGG Gag_180_209 0.034 34544.05 55.00
7 HLA-A0301 MLNTVGGH MLNTV-GGH 0 5 1 0 0 MLNTVGGH Gag_180_209 0.083 20462.88 19.00
8 HLA-A0301 LNTVGGHQ -LNTVGGHQ 0 0 1 0 0 LNTVGGHQ Gag_180_209 0.018 41270.38 85.00
9 HLA-A0301 NTVGGHQA NTVGGHQA- 0 8 1 0 0 NTVGGHQA Gag_180_209 0.015 42434.54 90.00
10 HLA-A0301 TVGGHQAA TVGGHQAA- 0 8 1 0 0 TVGGHQAA Gag_180_209 0.021 39642.67 80.00
11 HLA-A0301 VGGHQAAM -VGGHQAAM 0 0 1 0 0 VGGHQAAM Gag_180_209 0.021 39730.28 80.00
12 HLA-A0301 GGHQAAMQ GGHQAAMQ- 0 8 1 0 0 GGHQAAMQ Gag_180_209 0.015 42652.28 95.00
13 HLA-A0301 GHQAAMQM G-HQAAMQM 0 1 1 0 0 GHQAAMQM Gag_180_209 0.020 40135.11 80.00
14 HLA-A0301 HQAAMQML HQAAMQML- 0 8 1 0 0 HQAAMQML Gag_180_209 0.057 27116.52 31.00
15 HLA-A0301 QAAMQMLK -QAAMQMLK 0 0 1 0 0 QAAMQMLK Gag_180_209 0.238 3800.57 4.50
16 HLA-A0301 AAMQMLKE AAM-QMLKE 0 3 1 0 0 AAMQMLKE Gag_180_209 0.021 39659.42 80.00
17 HLA-A0301 AMQMLKET AMQMLKET- 0 8 1 0 0 AMQMLKET Gag_180_209 0.019 40509.00 85.00
18 HLA-A0301 MQMLKETI MQMLKET-I 0 7 1 0 0 MQMLKETI Gag_180_209 0.033 35088.76 60.00
19 HLA-A0301 QMLKETIN QMLKETIN- 0 8 1 0 0 QMLKETIN Gag_180_209 0.029 36469.85 65.00
20 HLA-A0301 MLKETINE MLKETINE- 0 8 1 0 0 MLKETINE Gag_180_209 0.027 37444.68 70.00
21 HLA-A0301 LKETINEE -LKETINEE 0 0 1 0 0 LKETINEE Gag_180_209 0.011 44465.09 99.00
22 HLA-A0301 KETINEEA KE-TINEEA 0 2 1 0 0 KETINEEA Gag_180_209 0.010 44649.25 99.00
0 HLA-A0301 TPQDLNTML TPQDLNTML 0 0 0 0 0 TPQDLNTML Gag_180_209 0.031 35876.13 60.00
1 HLA-A0301 PQDLNTMLN PQDLNTMLN 0 0 0 0 0 PQDLNTMLN Gag_180_209 0.029 36353.23 65.00
2 HLA-A0301 QDLNTMLNT QDLNTMLNT 0 0 0 0 0 QDLNTMLNT Gag_180_209 0.033 35061.82 60.00
3 HLA-A0301 DLNTMLNTV DLNTMLNTV 0 0 0 0 0 DLNTMLNTV Gag_180_209 0.056 27138.82 31.00
4 HLA-A0301 LNTMLNTVG LNTMLNTVG 0 0 0 0 0 LNTMLNTVG Gag_180_209 0.021 39713.52 80.00
5 HLA-A0301 NTMLNTVGG NTMLNTVGG 0 0 0 0 0 NTMLNTVGG Gag_180_209 0.043 31478.50 43.00
6 HLA-A0301 TMLNTVGGH TMLNTVGGH 0 0 0 0 0 TMLNTVGGH Gag_180_209 0.292 2129.03 3.00
7 HLA-A0301 MLNTVGGHQ MLNTVGGHQ 0 0 0 0 0 MLNTVGGHQ Gag_180_209 0.122 13419.03 11.00
8 HLA-A0301 LNTVGGHQA LNTVGGHQA 0 0 0 0 0 LNTVGGHQA Gag_180_209 0.021 39696.75 80.00
9 HLA-A0301 NTVGGHQAA NTVGGHQAA 0 0 0 0 0 NTVGGHQAA Gag_180_209 0.037 33383.30 49.00
10 HLA-A0301 TVGGHQAAM TVGGHQAAM 0 0 0 0 0 TVGGHQAAM Gag_180_209 0.078 21511.99 21.00
11 HLA-A0301 VGGHQAAMQ VGGHQAAMQ 0 0 0 0 0 VGGHQAAMQ Gag_180_209 0.020 40406.14 80.00
12 HLA-A0301 GGHQAAMQM GGHQAAMQM 0 0 0 0 0 GGHQAAMQM Gag_180_209 0.048 29872.45 38.00
13 HLA-A0301 GHQAAMQML GHQAAMQML 0 0 0 0 0 GHQAAMQML Gag_180_209 0.043 31303.24 42.00
14 HLA-A0301 HQAAMQMLK HQAAMQMLK 0 0 0 0 0 HQAAMQMLK Gag_180_209 0.681 31.42 0.15 <= SB
15 HLA-A0301 QAAMQMLKE QAAMQMLKE 0 0 0 0 0 QAAMQMLKE Gag_180_209 0.041 32014.67 45.00
16 HLA-A0301 AAMQMLKET AAMQMLKET 0 0 0 0 0 AAMQMLKET Gag_180_209 0.033 35022.77 60.00
17 HLA-A0301 AMQMLKETI AMQMLKETI 0 0 0 0 0 AMQMLKETI Gag_180_209 0.057 26947.74 31.00
18 HLA-A0301 MQMLKETIN MQMLKETIN 0 0 0 0 0 MQMLKETIN Gag_180_209 0.045 30830.30 41.00
19 HLA-A0301 QMLKETINE QMLKETINE 0 0 0 0 0 QMLKETINE Gag_180_209 0.064 25009.20 27.00
20 HLA-A0301 MLKETINEE MLKETINEE 0 0 0 0 0 MLKETINEE Gag_180_209 0.051 28662.32 35.00
21 HLA-A0301 LKETINEEA LKETINEEA 0 0 0 0 0 LKETINEEA Gag_180_209 0.013 43256.44 95.00
-----------------------------------------------------------------------------------
Protein Gag_180_209. Allele HLA-A0301. Number of high binders 1. Number of weak binders 0. Number of peptides 45
Link to Allele Frequencies in Worldwide Populations HLA-A0301
-----------------------------------------------------------------------------------
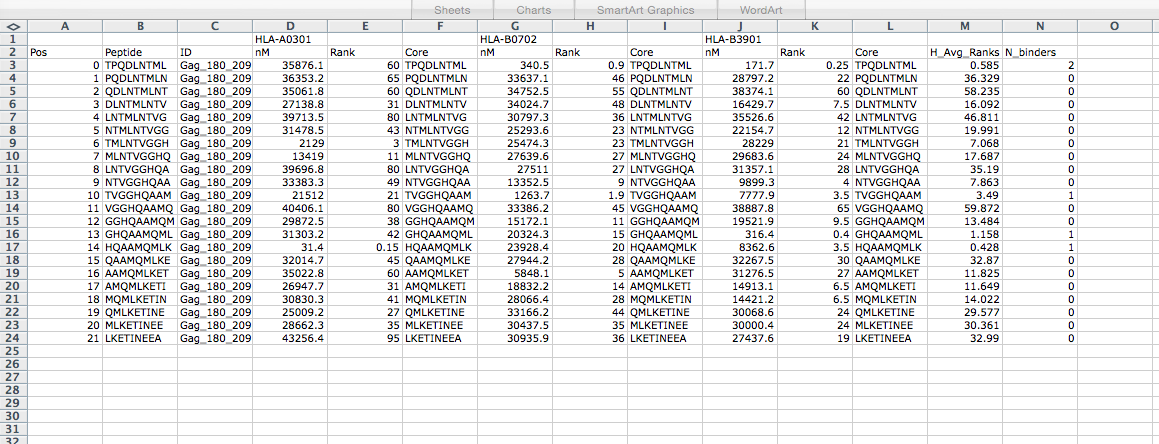
XLS output format
The XLS output summarizes the results in a convenient tabular format which can be read by spreadsheet programs such as Excel. For each peptide submitted for prediction, the table reports:
the predicted affinity (nM)
the percentile Rank, and
the predicted binding Core
for all the specified alleles.
Two additional columns summarize the predictions
across alleles. These quantitites may be useful for the design of peptides that ensure coverage across parts of the population:
H_Avg_Ranks harmonic mean of the %Rank calculated over all specified alleles
N_binders the number of alleles covered by a given peptide; in other words, how many MHC alleles the peptides is predicted to bind to.
Example XLS output:
References
Main references:
Gapped sequence alignment using artificial neural networks: application to the MHC class I system
Massimo Andreatta1 and Morten Nielsen1,2
Bioinformatics, Feb 15;32(4):511-7 2016
1Instituto de Investigaciones Biotecnológicas,
Universidad Nacional de San Martin,
San Martín, Buenos Aires, Argentina
2Center for Biological Sequence Analysis,
Technical University of Denmark,
DK-2800 Lyngby, Denmark
Motivation: Many biological processes are guided by receptor interactions with linear ligands of variable length. One such receptor is the MHC class I molecule. The length preferences vary depending on the MHC allele, but are generally limited to peptides of length 8 to 11 amino acids. On this relatively simple system, we developed a sequence alignment method based on artificial neural networks that allows insertions and deletions in the alignment.
Results: We show that prediction methods based on alignments that include insertions and deletions have significantly higher performance than methods trained on peptides of single lengths. Also, we illustrate how the location of deletions can aid the interpretation of the modes of binding of the peptide-MHC, as in the case of long peptides bulging out of the MHC groove or protruding at either terminus. Finally, we demonstrate that the method can learn the length profile of different MHC molecules, and quantified the reduction of the experimental effort required to identify potential epitopes using our prediction algorithm.
Availability: The NetMHC-4.0 method for the prediction of peptide-MHC class I binding affinity using gapped sequence alignment is publicly available at: http://www.cbs.dtu.dk/services/NetMHC-4.0.
Contact: mniel@cbs.dtu.dk
Supplementary information: Supplementary data are available at Bioinformatics online.
PMID: 26515819
[PDF]
NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11
Lundegaard C1Lamberth K2Harndahl M2Buus S2Lund O1Nielsen M1
Nucleic Acids Research 36 (suppl 2): W509-W512. 2008
1Center for Biological Sequence Analysis,
Technical University of Denmark,
DK-2800 Lyngby, Denmark
2Division of Experimental Immunology,
Institute of Medical Microbiology and Immunology,
University of Copenhagen, Denmark
NetMHC-3.0 is trained on a large number of quantitative peptide data using both affinity data from the Immune Epitope Database and Analysis Resource (IEDB) and elution data from SYFPEITHI. The method generates high-accuracy predictions of major histocompatibility complex (MHC): peptide binding. The predictions are based on artificial neural networks trained on data from 55 MHC alleles (43 Human and 12 non-human), and position-specific scoring matrices (PSSMs) for additional 67 HLA alleles. As only the MHC class I prediction server is available, predictions are possible for peptides of length 8–11 for all 122 alleles. artificial neural network predictions are given as actual IC50 values whereas PSSM predictions are given as a log-odds likelihood scores. The output is optionally available as download for easy post-processing. The training method underlying the server is the best available, and has been used to predict possible MHC-binding peptides in a series of pathogen viral proteomes including SARS, Influenza and HIV, resulting in an average of 75–80% confirmed MHC binders. Here, the performance is further validated and benchmarked using a large set of newly published affinity data, non-redundant to the training set.
PMID: 18463140
(full text version available)
Reliable prediction of T-cell epitopes using neural networks with novel
sequence representations.
Nielsen M1, Lundegaard C1, Worning P1,
Lauemoller SL2, Lamberth K2,Buus S2,
Brunak S1, Lund O1
Protein Sci., 12:1007-17, 2003.
1Center for Biological Sequence Analysis,
Technical University of Denmark,
DK-2800 Lyngby, Denmark
2Division of Experimental Immunology,
Institute of Medical Microbiology and Immunology,
University of Copenhagen, Denmark
In this paper we describe an improved neural network method to predict T-cell class I epitopes. A novel input representation has been developed consisting of a combination of sparse encoding, Blosum encoding, and input derived from hidden Markov models. We demonstrate that the combination of several neural networks derived using different sequence-encoding schemes has a performance superior to neural networks derived using a single sequence-encoding scheme. The new method is shown to have a performance that is substantially higher than that of other methods. By use of mutual information calculations we show that peptides that bind to the HLA A*0204 complex display signal of higher order sequence correlations. Neural networks are ideally suited to integrate such higher order correlations when predicting the binding affinity. It is this feature combined with the use of several neural networks derived from different and novel sequence-encoding schemes and the ability of the neural network to be trained on data consisting of continuous binding affinities that gives the new method an improved performance. The difference in predictive performance between the neural network methods and that of the matrix-driven methods is found to be most significant for peptides that bind strongly to the HLA molecule, confirming that the signal of higher order sequence correlation is most strongly present in high-binding peptides. Finally, we use the method to predict T-cell epitopes for the genome of hepatitis C virus and discuss possible applications of the prediction method to guide the process of rational vaccine design.
PMID: 2323871
(full text version available)
Sensitive quantitative predictions of peptide-MHC binding
by a 'Query by Committee' artificial neural network approach.
Buus S1, Lauemoller SL1, Worning P2,
Kesmir C2, Frimurer T2, Corbet S3,
Fomsgaard A3, Hilden J4, Holm A5,
Brunak S2.
Tissue Antigens., 62:378-84, 2003.
1Division of Experimental Immunology,
Institute of Medical Microbiology and Immunology,
University of Copenhagen, Denmark
2Center for Biological Sequence Analysis,
Technical University of Denmark,
DK-2800 Lyngby, Denmark
3Department of Virology,
State Serum Institute, Denmark
4Department of Biostatistics,
University of Copenhagen, Denmark
5Research Center for Medical Biotechnology,
Chemistry Department,
Royal Veterinary and Agricultural University, Denmark
We have generated Artificial Neural Networks (ANN) capable of performing
sensitive, quantitative predictions of peptide binding to the MHC class I
molecule, HLA-A*0204. We have shown that such quantitative ANN are superior to
conventional classification ANN, that have been trained to predict binding vs
non-binding peptides. Furthermore, quantitative ANN allowed a straightforward
application of a 'Query by Committee' (QBC) principle whereby particularly
information-rich peptides could be identified and subsequently tested
experimentally. Iterative training based on QBC-selected peptides considerably
increased the sensitivity without compromising the efficiency of the
prediction. This suggests a general, rational and unbiased approach to the
development of high quality predictions of epitopes restricted to this and
other HLA molecules. Due to their quantitative nature, such predictions will
cover a wide range of MHC-binding affinities of immunological interest, and
they can be readily integrated with predictions of other events involved in
generating immunogenic epitopes. These predictions have the capacity to perform
rapid proteome-wide searches for epitopes. Finally, it is an example of an
iterative feedback loop whereby advanced, computational bioinformatics optimize
experimental strategy, and vice versa.
PMID: 14617044
(full text version available)