Instructions
1. Specify the input sequences
All the input sequences must be in one-letter amino acid
code. The allowed alphabet (not case sensitive) is as follows:
A C D E F G H I K L M N P Q R S T V W Y and X (unknown)
All the alphabetic symbols not in the allowed alphabet
will be converted to X before processing. All the non-alphabetic
symbols, including white space and digits, will be ignored.
The sequences can be input in the following two ways:
-
Paste a single sequence (just the amino acids) or a number of sequences in
FASTA
format into the upper window of the main server page.
-
Select a FASTA
file on your local disk, either by typing the file name into the lower window
or by browsing the disk.
Both ways can be employed at the same time: all the specified sequences will
be processed. However, there may be not more than 5,000 sequences in one submission. The sequences
may not be longer than 10,000 amino acids.
2. Customize your run
Generating figures for a large number of samples takes much longer than
executing a prediction. Consider using the short option for large sample
batches.
- Output format:
You can choose between two output formats:
- Long
- Appropriate for most users. Shows one plot and one summary per sequence.
- Short
- Convenient if you submit lots of sequences. Shows only one line of
output per sequence and no graphics.
3. Submit the job
Click on the
"Submit" button. The status of your job (either 'queued'
or 'running') will be displayed and constantly updated until it terminates and
the server output appears in the browser window.
At any time during the wait you may enter your e-mail address and simply leave
the window. Your job will continue; you will be notified by e-mail when it has
terminated. The e-mail message will contain the URL under which the results are
stored; they will remain on the server for 24 hours for you to collect them.
Example Outputs
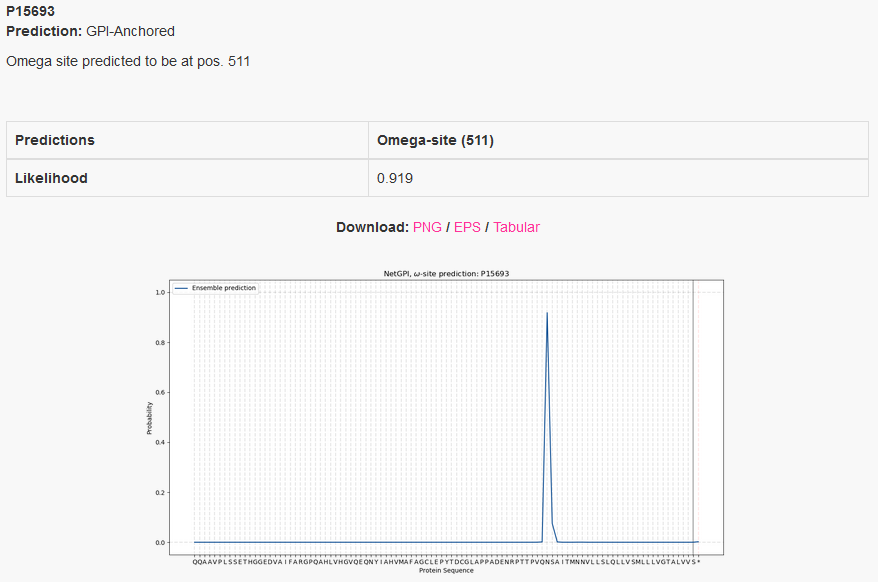
By default the server produces the following output for each input sequence.
The example below shows the output for intestinal-type alkaline phosphatase 1,
taken from the
Uniprot entry
PPBI1_RAT. The lipidation
position prediction is consistent with the database annotation.
One annotation is attributed to each protein, the one that has the highest
probability. If the highest probability is within the amino-acid sequence, then
it is considered GPI-anchored and the amino-acid position at the peak is the
predicted omega-site. If the highest probability is at the sentinel,
here represented by *, then the protein is considered non GPI-anchored.
If a GPI-anchor is predicted, the omega-site position is reported as well.
On the plot we see the likelihood distribution over the protein sequence, with
the added sentinel *. Only the last 100 amino-acids are considered.
Example: Mature protein - standard output format
The NetGPI dataset
The datasets for training and benchmarking NetGPI-1.1 can be found here. The dataset is provided in 2-line FASTA format.
The format is as follows:
>uniprot_ac|kingdom|anchoring|pos_from_end|pos_from_beginning|part_no|anchor_exp|omega_exp
amino-acid sequence
where:
- uniprot_ac is an accession number
- kingdom is the organism's kingdom
- anchoring is GPI-anchored or non_GPI-anchored
- pos_from_end is the position within the sequence from the end, where 0 is the sentinel
- pos_from_beginning is the position within the truncated sequence from the beginning
- part_no is the partition that the protein is assigned to
- anchor_exp is 1 when the entry has experimental evidence for the GPI-anchoring signal sequence, 0 otherwise
- omega_exp is 1 when the entry has experimental evidence for the omega-site, 0 otherwise
NetGPI dataset: download
Article abstract
Current version (NetGPI v. 1.1)
Prediction of GPI-anchored proteins with pointer neural networks
Magnús Halldór Gíslason, Henrik Nielsen, José Juan Almagro Armenteros, Alexander Rosenberg Johansen.
GPI-anchors constitute a very important post-translational modification, linking many proteins to the outer face of the plasma membrane in eukaryotic cells.
Since experimental validation of GPI-anchoring signals is slow and costly, computatinal approaches for predicting them from amino acid sequences are needed.
However, the most recent GPI predictor is more than a decade old and considerable progress has been made in machine learning since then.
We present a new dataset and a novel method, NetGPI, for GPI signal prediction. NetGPI is based on recurrent neural networks, incorporating an attention mechanism that simultaneously detects GPI-anchoring signals and points out the location of their ω-sites.
The performance of NetGPI is superior to existing methods with regards to discrimination between GPI-anchored proteins and other secretory proteins and approximate (±1 position) placement of the ω-site.
Current Research in Biotechnology, 3, 6-13, doi: https://doi.org/10.1016/j.crbiot.2021.01.001 (2021)