Submission
CITATIONS
For publication of results, please cite:
- Current version:
NetCTL-1.2:
Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction.
Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M.
BMC Bioinformatics. Oct 31;8:424. 2007
View the abstract
- Earlier versions:
NetCTL-1.0:
An integrative approach to CTL epitope prediction.
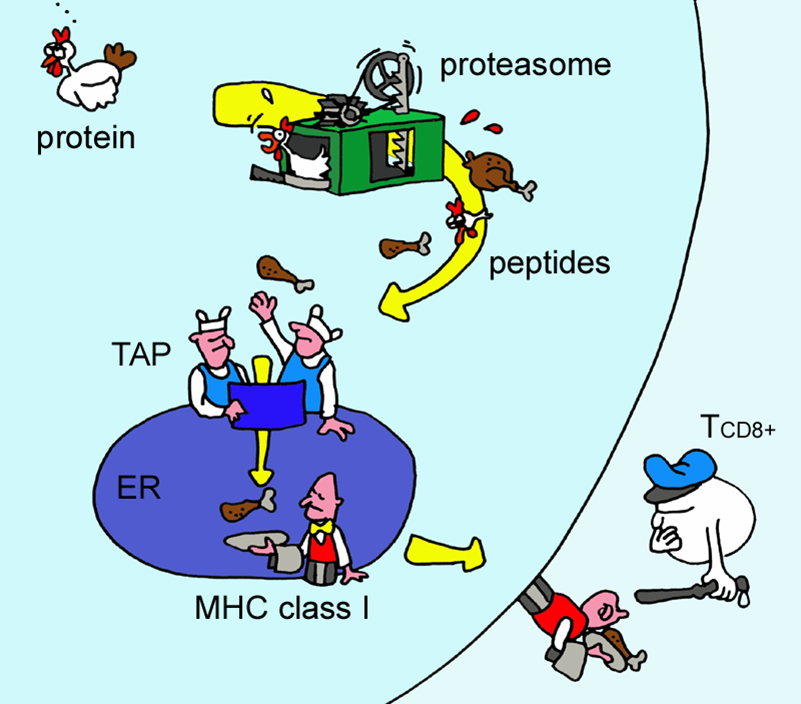
A combined algorithm integrating MHC-I binding, TAP transport efficiency, and proteasomal cleavage predictions.
Larsen M.V., Lundegaard C., Kasper Lamberth, Buus S,. Brunak S., Lund O., and Nielsen M.
European Journal of Immunology. 35(8): 2295-303. 2005
View the abstract
- Related publications:
Reliable prediction of T-cell epitopes using neural networks with novel
sequence representations.
Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S,
Brunak S, Lund O.
Protein Sci., 12:1007-17, 2003.
View the abstract
Sensitive quantitative predictions of peptide-MHC binding by a 'Query by Committee' artificial neural network approach.
Buus S, Lauemoller SL, Worning P, Kesmir C, Frimurer T, Corbet S, Fomsgaard A, Hilden J, Holm A, Brunak S.
Tissue Antigens., 62(5):378-84, 2003.
View the abstract
The role of the proteasome in generating cytotoxic T cell epitopes:
Insights obtained from improved predictions of proteasomal cleavage.
M. Nielsen, C. Lundegaard, S. Brunak, O. Lund, and C. Kesmir.
Immunogenetics., 57(1-2):33-41, 2005.
View the abstract
Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors
Peters, B., Bulik, S., Tampe, R., Endert, P. M. V. and Holzhutter, H. G.
J. Immunol. 171: 1741-1749, 2003.
View the abstract
Instructions
In order to use the NetCTL server for prediction on amino acid sequences:
- Enter the sequence in the sequence window, or give a file name.
The sequence must be written using the one letter
amino acid code:
`acdefghiklmnpqrstvwy' or
`ACDEFGHIKLMNPQRSTVWY'.
Other letters will be converted to `X' and treated as unknown
amino acids.
Other characters, such as whitespace and
numbers, will simply be ignored.
- Select HLA supertype (10 HLA supertypes are available)
- Optional
- Weight on C terminal cleavage. The default select value (0.1) gives optimal predictive performance on
average. You can modify the relative weight on proteasomal cleavage by entering a different weight value.
- Weight on TAP transport efficiency. The default select value (0.05) gives optimal predictive performance on
average. You can modify the relative weight on TAP transport by entering a different weight value.
- Threshold for epitope identification. Peptides with a combined prediction score value greater than the
threshold value are marked as potential epitopes. In a large scale benchmark identifying known CTL
epitope in proteins the default value of 0.75 was found to correspond to a sensitivity of 0.65 and a
specificity 0.97. Note that the benchmark is highly unbalanced since only one peptide is identified as CTL
epitope in each protein, and the number of negatives hence is orders of magnitude larger than the number of
positives. This has important implications for the interpretation of the specificity values.
- Sort by score. Select which feature to sort on. Sorting is done in decreasing order. Default is no
sorting.
- Press the "Submit sequence" button.
- A WWW page will return the results when the prediction is ready.
Response time depends on system load.
Output format
Description
The output consists of 16 columns:
Residue number.
ID
Protien identifier
pep
Peptide sequence
aff
Predicted MHC binding affinity. The value is give as 1 - log50k(aff), where log50k is the logaritm with base 50.000, and aff is the affinity in nM units
aff_rescale
Rescale binding affinity. The predicted binding affinity is normalized by the 1st percentile score
cle
C terminal cleavage affinity
tap
TAP transport efficiency
COMB
Prediction score
<-E. Identified MHC ligands
EXAMPLE OUTPUT
NetCTL predictions using MHC supertype A24. Threshold 0.500000
.......
17 ID 143B_BOVIN pep AERYDDMAA aff 0.0349 aff_rescale 0.0478 cle 0.0344 tap -0.5270 COMB 0.0249
18 ID 143B_BOVIN pep ERYDDMAAA aff 0.0348 aff_rescale 0.0476 cle 0.4333 tap -0.1920 COMB 0.0813
19 ID 143B_BOVIN pep RYDDMAAAM aff 0.3017 aff_rescale 0.4128 cle 0.9943 tap 0.4700 COMB 0.5357 <-E
20 ID 143B_BOVIN pep YDDMAAAMK aff 0.0352 aff_rescale 0.0482 cle 0.9622 tap 0.0930 COMB 0.1491
21 ID 143B_BOVIN pep DDMAAAMKA aff 0.0343 aff_rescale 0.0469 cle 0.6469 tap -1.1460 COMB 0.0543
......
----------------------------
Number of MHC ligands 8 identified. Number of amino acids 237. Protein name 143B_BOVIN
----------------------------
Supplementary material for update to the NetCTL method (v. 1.2):
Original method
An integrative approach to CTL epitope prediction.
A combined algorithm integrating MHC-I binding, TAP transport efficiency, and proteasomal cleavage predictions.
The two data sets used in the benchmark calculation are given below in the FASTA format. For each entry is given the
Swiss-Prot name of the protein "hosting" the epitope, the epitope position,
the epitope sequence, and the HLA supertype.
SYFPEITHI dataset
HIV dataset
HIV_EpiJen dataset
List of known CTL epitopes from SYFPEITHI and the Los Alamos HIV databases
Difference between the A1, A2, and A3 epitope-protein pairs for the HIV and HIV_EpiJen datasets
Supplementary figure: Comparing specificities on the HIV EpiJen dataset
References
M. V. Larsen, C. Lundegaard, K. Lamberth, S. Buus, S. Brunak, O. Lund, and M. Nielsen
An integrative approach to CTL epitope prediction. A combined algorithm integrating MHC binding,
TAP transport efficiency, and proteasome cleavage predictions.
European Journal of Immunology 2005 Aug;35(8):2295-303.
Abstract
Reference
Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction.
Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M.
BMC Bioinformatics. 2007 Oct 31;8:424.
Abstract
BACKGROUND: Reliable predictions of Cytotoxic T
lymphocyte (CTL) epitopes are essential for rational vaccine design.
Most importantly, they can minimize the experimental effort needed to
identify epitopes. NetCTL is a web-based tool designed for predicting
human CTL epitopes in any given protein. It does so by integrating
predictions of proteasomal cleavage, TAP transport efficiency, and MHC
class I affinity. At least four other methods have been developed
recently that likewise attempt to predict CTL epitopes: EpiJen, MAPPP,
MHC-pathway, and WAPP. In order to compare the performance of
prediction methods, objective benchmarks and standardized performance
measures are needed. Here, we develop such large-scale benchmark and
corresponding performance measures and report the performance of an
updated version 1.2 of NetCTL in comparison with the four other
methods.
RESULTS: We define a number of performance measures that can
handle the different types of output data from the five methods. We use
two evaluation datasets consisting of known HIV CTL epitopes and their
source proteins. The source proteins are split into all possible 9 mers
and except for annotated epitopes; all other 9 mers are considered
non-epitopes. In the RANK measure, we compare two methods at a time and
count how often each of the methods rank the epitope highest. In
another measure, we find the specificity of the methods at three
predefined sensitivity values. Lastly, for each method, we calculate
the percentage of known epitopes that rank within the 5% peptides with
the highest predicted score.
CONCLUSION: NetCTL-1.2 is demonstrated to
have a higher predictive performance than EpiJen, MAPPP, MHC-pathway,
and WAPP on all performance measures. The higher performance of
NetCTL-1.2 as compared to EpiJen and MHC-pathway is, however, not
statistically significant on all measures. In the large-scale benchmark
calculation consisting of 216 known HIV epitopes covering all 12
recognized HLA supertypes, the NetCTL-1.2 method was shown to have a
sensitivity among the 5% top-scoring peptides above 0.72. On this
dataset, the best of the other methods achieved a sensitivity of 0.64.
The NetCTL-1.2 method is available at
http://services.healthtech.dtu.dk/service.php?NetCTL
All used datasets are available
at http://services.healthtech.dtu.dk/suppl/immunology/CTL-1.2.php