Description of data sets
Dataset extraction
Homology reduction
Sequence logos
Download the training sets
Data set
The section describes the extraction and homology reduction of the data sets used for training of NetAcet 1.0.Extraction
The data used for NetAcet were extracted
from Table 2 in Polevoda et al. and from the Yeast Protein Map.
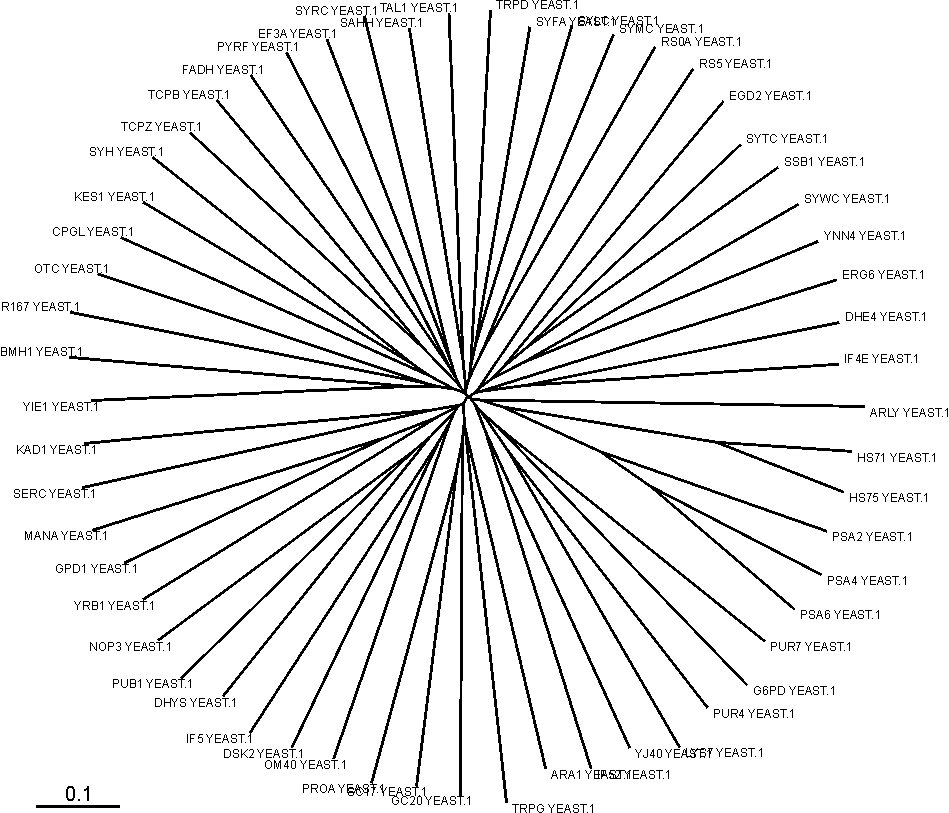
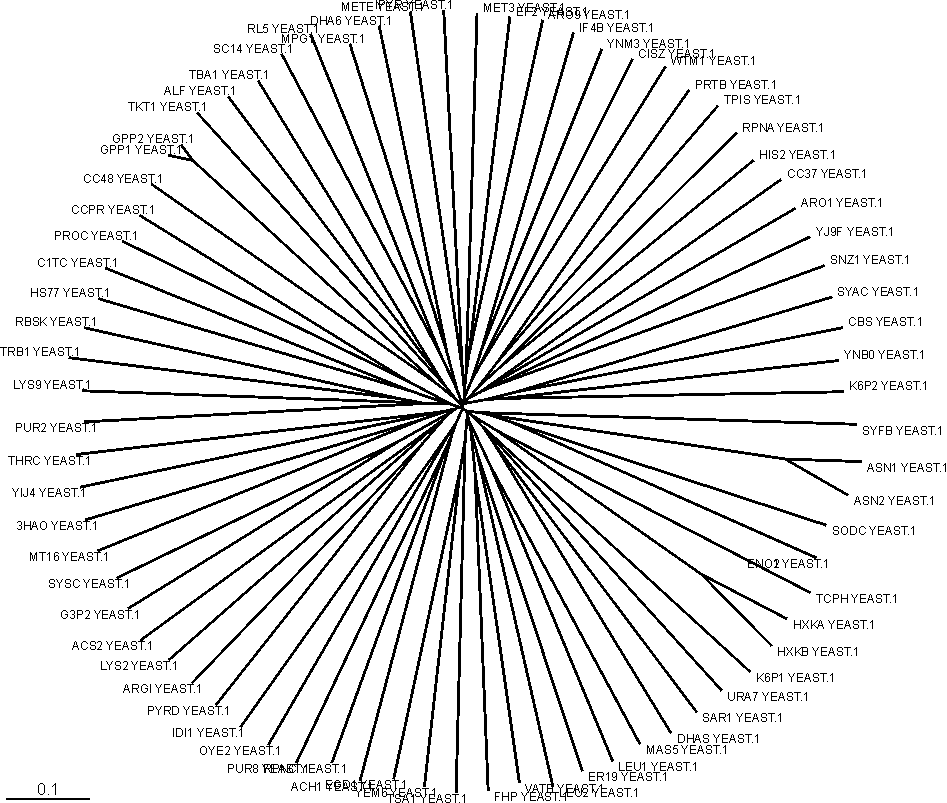
All inconsistensies between the two data sets were removed resulting in a positive set of 61 sequences and 76 negative sequences.
Sequences were truncated to their N-terminal 40 residues and subsequently homology
reduced by visual inspection of a neighbour-joining tree generated from a ClustalW
multible alignment. Four sequences were removed from the positive dataset due to close
homology to other sequences and following this reduction the two closest homologs were 52% identical although the average homology is much lower.
Below is shown an unrooted phylogenetic tree of the positive data set before homology reduction.
Below is shown an unrooted phylogenetic tree of the negative data set before homology reduction.
To visualise the sequence information content for N-terminal acetylation,
we have generated sequence logos for the yeast training set.
The total height of the stack of letters at each position shows
the amount of sequence conservation at the position, while the relative
height of each letter shows the relative abundance of the corresponding
amino acid.
Homology reduction
Sequence logos
![]()

Blue:
Positively charged residues
Red:
Negatively charged residues
Green:
Neutral polar residues
Black:
Hydrophobic residues
Download the dataset
The datasets used for the training of NetAcet can be downloaded here:
Positive training set
Negative training set