Introduction
Contents
Background and Motivation
MatrixPlot is a program to display high quality matrix plots of any type
of data given in a simple format, the .mp format. MatrixPlot have a number of
options to tune the plot according to specifications given by the user. In
particular the user may display additional information along the edges of the
plot and zoom in on any region of the plot. For further introduction consult
the MatrixPlot paper
MatrixPlot: a program to visualize sequence constraints.
J. Gorodkin, H. H. Stærfeldt, O. Lund, and S. Brunak.
Bioinformatics. 15:769-770, 1999. (http://www.cbs.dtu.dk/services/MatrixPlot/)
MatrixPlot is accompanied by two other programs:
Inform, which computes
the mutual information between any two positions in a sequence alignment, and
produces the output in the mp format. The second program
pdb2mp takes a
PDB file from
Brookhaven Protein Data Bank and computes all
the interatomic distance between either the C-alpha atoms in proteins or P
atoms in nucleotide sequences. The output is produced in the mp format. The
modularity of the program structure is simple:
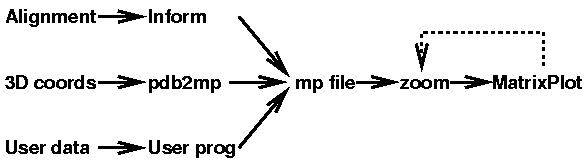 In particular for the user program, one can export the output of any web server
directly to MatrixPlot by using hidden variables in a submission form. The zoom
program cuts out a desired region of the matrix plot. It can be reused
arbitrarily many times on the mp file typically after consulting the output
from MatrixPlot.
In particular for the user program, one can export the output of any web server
directly to MatrixPlot by using hidden variables in a submission form. The zoom
program cuts out a desired region of the matrix plot. It can be reused
arbitrarily many times on the mp file typically after consulting the output
from MatrixPlot.
The mutual information content of a structural alignment (refound by foldalign of
the following RNA sequences (sequences from Tuerk et al. PNAS 89, pp
6988-6992, 1992.):
CCAGAGGCCCAACUGGUAAACGGGC
CCG-AAGCUCAACGGGAUAAUGAGC
CCG-AAGCCGAACGGGAAAACCGGC
CC-CAAGCGC-AGGGGAGAA-GCGC
CCG-ACGCCA-ACGGGAGAA-UGGC
CCGUUUUCAG-UCGGGAAAAACUGA
CCGUUACUCC-UCGGGAUAAAGGAG
CCGUAAGAGG-ACGGGAUAAACCUC
CCG-UAGGAG-GCGGGAUAU-CUCC
CCG--UGCCG-GCGGGAUAU-CGGC
CCG-AACUCG-ACGGGAUAA-CGAG
CCG--ACUCG--CGGGAUAA-CGAG
|
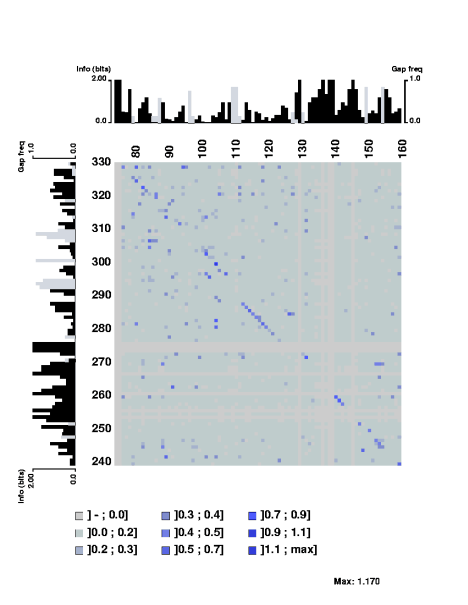
can be displayed by MatrixPlot as:
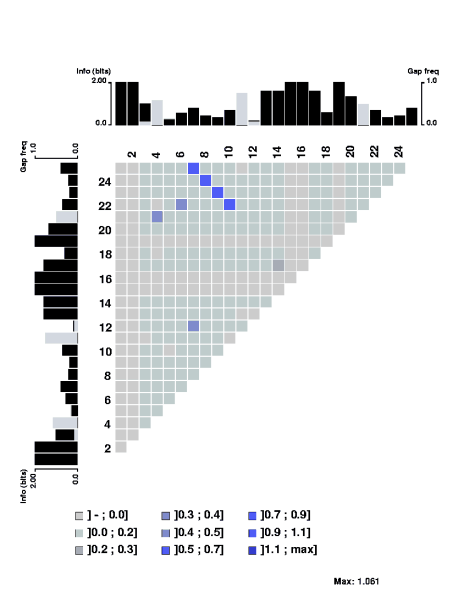 The degree of mutual information is indicated by the color scale. Below we
discuss convenient ways to compute the mutual information content of such a
structural alignment. Along the edges the plain sequence information content
along with gap frequencies are displayed. The scale to the left of the profile
indicates the sequence information, which is plotted in black. The scale to the
right indicates the frequency of gaps. The sequence information is computed as
in the structure
logo, but when including gaps we have introduced displacement of the
baseline of the plain sequence information content profile. This is discussed
below. If, at some position in the alignment, the gap frequency uses a larger
bar than than for the sequence information content, the information bar is
displayed on top of the gap bar, and vice versa.
The degree of mutual information is indicated by the color scale. Below we
discuss convenient ways to compute the mutual information content of such a
structural alignment. Along the edges the plain sequence information content
along with gap frequencies are displayed. The scale to the left of the profile
indicates the sequence information, which is plotted in black. The scale to the
right indicates the frequency of gaps. The sequence information is computed as
in the structure
logo, but when including gaps we have introduced displacement of the
baseline of the plain sequence information content profile. This is discussed
below. If, at some position in the alignment, the gap frequency uses a larger
bar than than for the sequence information content, the information bar is
displayed on top of the gap bar, and vice versa.
For sequences with a set of three-dimensional coordinates, for example PDB
entry 1raa chain A, MatrixPlot generates the plot
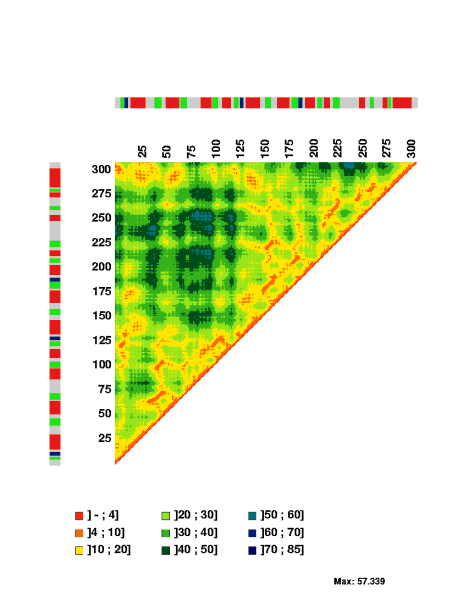 The interatomic distances between the C-alpha atoms are indicated by the
colorscales, which measures distances in Angstrom. The colors in the bar
express the secondary structure assignment for the PDB file: helix is indicated
in red; sheet is indicated in green; turn is indicated in blue. The color gray
indicate residues without assignment in the PDB file.
The interatomic distances between the C-alpha atoms are indicated by the
colorscales, which measures distances in Angstrom. The colors in the bar
express the secondary structure assignment for the PDB file: helix is indicated
in red; sheet is indicated in green; turn is indicated in blue. The color gray
indicate residues without assignment in the PDB file.
Below it is discussed how to modify the graphical output of MatrixPlot.
The graphical output is given in postscript files. Any submitted data is kept
confidential and deleted a short time after submission.
Data Formats
- Inform formats:
The formats for Inform are those that can be used for the MatrixPlot
pages for generating mutual information plots of
nucleotide sequence and
proteins. The formats are
a simple align format, fasta format and the msf format. Examples:
- pdb2mp formats
- MatrixPlot format
Program Options (man pages)
The options to Inform, pdb2mp, MatrixPlot, and Zoom are described in the pages
given below. Source code requests: send email to gorodkin@cbs.dtu.dk.
WWW Interface
The web interface of MatrixPlot consist of four pages, and has been made from
the programs presented above: mutual information plots for RNA or DNA
alignments, mutual information plots for protein alignments, distance matrices
of sequences with a set of three-dimensional coordinates, and a page that takes
a user produced mp file. Some of the command line options of the individual
programs have been combined into one field on the corresponding web page. This
is described here:
-
Mutual information for nucleotide
sequence alignments
This page contains the options to Inform and all the relevant
options of MatrixPlot to generate mutual information plots.
-
Mutual information for protein sequence
alignments
This page contains the options to Inform ("complementarity
matrix" is not defined) with mtype=1 (standard form of mutual information), and
all the relevant options of MatrixPlot.
-
Distance matrices
This page contains the options to pdb2mp, as well as the relevant MatrixPlot
options to generate distance matrices of sequences with a set of 3D
coordinates. The user may also enter the name of a PDB entry. If the chain
identifier is omitted a distance matrix will be generated for the first
identifier in the PDB entry.
-
User matrix
This page allows the user to submit an mp file. All MatrixPlot options are
available on that page.
After submission to the web pages, the matrix plot is returned as gif and
postscript files. The colors on the gif file are limited, so they might be
distorded depending on the browser. Along with the matrix plots a new form is
given. This form includes all the previous MatrixPlot settings as well as
options to zoom around in the plot. Upon resubmission new matrix plots are
returned together with a new submission form. The following two plots
illustrate this. First a full matrix plot of the archaea RNA sequences
downloaded from the Signal recognition particle database (Samuelsson and Zwieb, Nucl. Acids Res. 27 pp
169-170, 1999). The trace of lines from upper left corner towards lower right
corner indicate strecthes of covariant positions in the alignment.
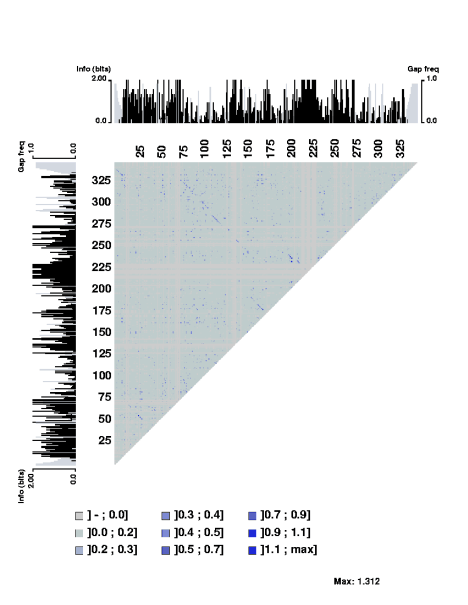 Then using the zoom option result in the following plot:
Then using the zoom option result in the following plot:
The stretch of covarying positions corresponds to the main stem of the RNA
secondary structure. Note that the trace is interrupted by a few positions
having complete sequence conservation. The respective parts of the sequence
logo profile have also been displayed along the edges of the plot.
Export user data directly to MatrixPlot
The user data can automatically be exported to MatrixPlot thorugh the
web. This would typically be data generated from another web page which then
can be exported for graphical manipulation, such as zooming in on a region.
Only a single button is needed, as the form can be written using hidden
variables.
Discussion of Information Content
Two issues on information content for sequence/structure alignments are
discussed here. First the contribution to the computation of information
content when gaps are included in the alignment, and secondly the sample size
robustness of mutual information for RNA secondary structure co-variance
analysis. For background and details consult the mentioned references.
-
Calculating the information content of an alignment that have gaps.
The conceptual problem in computing the information content or relative entropy
of an alignment that has gaps is the background probablities. The background
probabilities are typically found by counting base (or amino acid) frequencies
in the genomes (or data set) from which the aligned sequences originate. The
gaps, however, do not appear before the alignment is made, so a ``gap
background probability'' or a ``gap expectation value'' cannot be dealt with in
the same way. It only makes sense to talk about this when the alignment has
been performed.
Calculation of the information content of an alignment containing gaps has been
derived by Hertz and Stormo, 1995 (In Lim & Cantor (eds), Proc. of the
Third Int. Conf. on Bioinformatics and genome Res., pp. 201-216). They derived
the expression
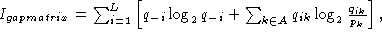 where
where 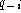 and
and 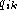 are the fraction
of gaps and symbol k at position i in the alignment, and where
k is a symbol in the alphabet A. The fraction
are the fraction
of gaps and symbol k at position i in the alignment, and where
k is a symbol in the alphabet A. The fraction 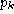 is the background probability of symbol
k, so
is the background probability of symbol
k, so 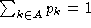 . Note that if
only a few sequences contain a gap (
. Note that if
only a few sequences contain a gap (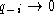 ) or almost all
sequences have a gap (
) or almost all
sequences have a gap (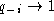 ) at some position
i, the ``gap'' term does not contribute to the sum. Since the gap
background probability is one in this expression, then all the background
probabilities sum to 2 rather than one, which is the formal claim to define
information content or relative entropy. This is resolved as follows. Hertz and
Stormo derived a large-devation rate function given by
) at some position
i, the ``gap'' term does not contribute to the sum. Since the gap
background probability is one in this expression, then all the background
probabilities sum to 2 rather than one, which is the formal claim to define
information content or relative entropy. This is resolved as follows. Hertz and
Stormo derived a large-devation rate function given by
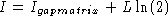 and as they write, it normalizes all background probabilities
with a factor of 1/2. This corresponds to adding
and as they write, it normalizes all background probabilities
with a factor of 1/2. This corresponds to adding
 to the
information content on each of L positions in the alignment. Clearly,
the difference between I and
to the
information content on each of L positions in the alignment. Clearly,
the difference between I and 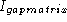 , is where to put
the baseline in a sequence logo profile plot. The normalizing factor of
Hertz and Stormo was found
by considering the minimum of .
When using the normalizing factor, one interpretation is that the a
priori expectation to gaps is probability 0.5, however, another
interpretation is that one has two models, one for which the expectation values
for all the symbols sum to one, and one model dealing only with gaps and the
expectation to gaps is one. When combining the two models a (re-)normalization
of all the parameters can be introduced.
, is where to put
the baseline in a sequence logo profile plot. The normalizing factor of
Hertz and Stormo was found
by considering the minimum of .
When using the normalizing factor, one interpretation is that the a
priori expectation to gaps is probability 0.5, however, another
interpretation is that one has two models, one for which the expectation values
for all the symbols sum to one, and one model dealing only with gaps and the
expectation to gaps is one. When combining the two models a (re-)normalization
of all the parameters can be introduced.
The considerations for any gap background probability can in general be
extended by subtracting from the standard
form
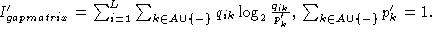 When using the normalization
When using the normalization 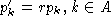 , it is easily
found that
, it is easily
found that 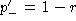 . Clearly
. Clearly 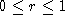 . Hence, the
normalizing factor and the a priori expectation of gap frequency is
directly related and the same when
. Hence, the
normalizing factor and the a priori expectation of gap frequency is
directly related and the same when 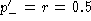 . Using this
relation one can readily write
. Using this
relation one can readily write
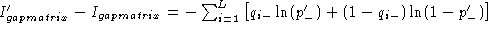 so when the difference is equal to
so when the difference is equal to 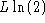 , and the same as above is obtained. The result
becomes independent of . The
interpretation of a gap a priori probability of 0.5 also makes sense.
If no prior knowledge is known about the alignment it makes sense to expect
gaps to appear randomly, i.e. having an a priori expectation of 0.5.
So a ``renormalization'' of the two sets of probabilities, the set of base
frequencies, and the gap probability, ensures that the background probabilities
sum to one and fulfill the formal claim to define information content.
, and the same as above is obtained. The result
becomes independent of . The
interpretation of a gap a priori probability of 0.5 also makes sense.
If no prior knowledge is known about the alignment it makes sense to expect
gaps to appear randomly, i.e. having an a priori expectation of 0.5.
So a ``renormalization'' of the two sets of probabilities, the set of base
frequencies, and the gap probability, ensures that the background probabilities
sum to one and fulfill the formal claim to define information content.
The current version of Inform computes the information content according to
, but MatrixPlot contains an option ``neg'' for
which the baseline can be moved as described (when neg=n). Otherwise (when
neg=y) the baseline is placed so that ``negative information content'' can show
up on the profile. This can be useful in identifying positions which neither
contains many or few gaps, but really is degenerated.
-
Mutual information only for basepairs
As has been discussed by Gorodkin et al. (Comput. Appl. Biosci.,
13:583-586, 1997) and used in the display of structure logos the
mutual information content for RNA sequences can be limited to include only
pairs of bases that are complementary. In that way a more relevant measure is
constructed. This is done by defining a complementary matrix (Gorodkin
et al. Nucl. Acids. Res. 25:3724-3732,1997) which lists the bases that
are complementary. The complementary matrix lists all bases against themselves,
and a number is assigned to indicate the degree of ``belief'' in the
complementarity. The matrix is clearly symmetric and most positions in the
matrix hold the value zero. The mutual information between position i
and j in the alignment using the complementary matrix is given by
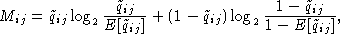 (A)
where
(A)
where 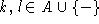 , A the alphabet, and where
, A the alphabet, and where 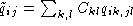 and
and 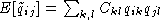 . The element
. The element 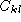 of the complementary matrix
lists the degree of complementarity between base k and l. The
fraction of pairs of base k at position i and base l at
position j is
of the complementary matrix
lists the degree of complementarity between base k and l. The
fraction of pairs of base k at position i and base l at
position j is 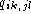 and
and 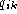 is the
fraction of base k at position i. Gaps is dealt with by having
is the
fraction of base k at position i. Gaps is dealt with by having
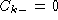 for any base k.
There are in particular two qualitative differences between this measure
and the standard form of mutual
information given by
for any base k.
There are in particular two qualitative differences between this measure
and the standard form of mutual
information given by
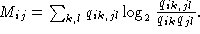 (B)
First in (A) the observed and expected values are computed independently for
two positions i and j, and the the overall covariance is
computed. In contrast in (B) the observed and expected values are compared for
each combination of the involved symbols. Secondly, in (A) only symbols for
which a basepair rule is defined are included in the sum. In (B) all symbols
are included (also gaps) even if it is already known that they do not (for RNA
secondary structure) contribute to the covariance. This fact makes (A) much
more robust to sampling size noise as illustrated below by two examples. In the
Inform program measure (A) is referred to as mtype 2, and (B) to mtype 1.
The upper triangle is mutual information content computed by type 2, and
the lower triangle is the mutual information content computed by using type 1.
(B)
First in (A) the observed and expected values are computed independently for
two positions i and j, and the the overall covariance is
computed. In contrast in (B) the observed and expected values are compared for
each combination of the involved symbols. Secondly, in (A) only symbols for
which a basepair rule is defined are included in the sum. In (B) all symbols
are included (also gaps) even if it is already known that they do not (for RNA
secondary structure) contribute to the covariance. This fact makes (A) much
more robust to sampling size noise as illustrated below by two examples. In the
Inform program measure (A) is referred to as mtype 2, and (B) to mtype 1.
The upper triangle is mutual information content computed by type 2, and
the lower triangle is the mutual information content computed by using type 1.
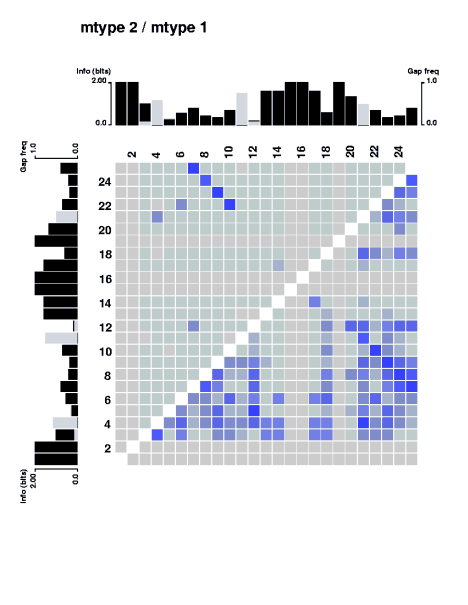
Sample size corrections for plain sequence information content (e.g.
Schneider et al. J. Mol. Biol. 188:415-431, 1986; Basharin, Theory
Probability Appl. 4:333-336, 1959), and for the standard form of mutual
information and correlation functions (e.g. Weiss and Herzel, J.
Theor. Biol. 190:341-353, 1998) has previously been studied.
Acknowledgements
Thanks to Anders Krogh for his contribution in the discussion of information
content. Thanks to Claus A. Andersen, Lars J. Jensen, and Christopher Workman
for suggestions and feedback.
Reference
MatrixPlot: visualizing sequence constraints.
J. Gorodkin, H. H. Stærfeldt, O. Lund, and S. Brunak.
Bioinformatics. 15:769-770, 1999. (http://www.cbs.dtu.dk/services/MatrixPlot/)
Data formats
This page is an individual page for the data format, and can also be found
as a section on the introduction page. The data formats used by the programs,
Inform, pdb2mp, and MatrixPlot are described and illustrated by examples.
- Inform formats:
The formats for Inform are those that can be used for the MatrixPlot
pages for generating mutual information plots of
nucletide sequences and proteins. The formats are
a simple align format, fasta format and the msf format. Examples:
- pdb2mp formats
- MatrixPlot format
