CITATIONS
For publication of results, please cite:
-
Accurate MHC Motif Deconvolution of immunopeptidomics data reveals high relevant contribution of DRB3, 4 and 5 to the total DR Immunopeptidome.
Saghar Kaabinejadian, Carolina Barra, Bruno Alvarez, Hooman Yari, William Hildebrand, Morten Nielsen
Frontiers in Immunology 26 January 2022. Sec. Antigen Presenting Cell Biology, DOI: 10.3389/fimmu.2022.835454
DATA RESOURCES
###################################################
# Running MHC_Motif_Decon 1.0 ...
#
# Call from /var/www/webface/tmp/server/mhcmotifdecon-1.0/61DCAF1F000013A1AED09789
#
# Input file: /var/www/webface/tmp/server/mhcmotifdecon-1.0/61DCAF1F000013A1AED09789/file.0
# Number of sequences: 100
# Label-MHC file: /var/www/webface/tmp/server/mhcmotifdecon-1.0/61DCAF1F000013A1AED09789/file.1
# MHC class: II
# Length range: 12-21
# RANK threshold: 20
# Minimum quantity of sequences for logo plotting: 10
# MHC counts plots will be included.
# MHC length histograms will be included.
# Run ID: run_21019
# Dirty mode enabled.
#
# Creating output folder /var/www/webface/tmp/server/mhcmotifdecon-1.0/61DCAF1F000013A1AED09789/run_21019...
# DONE.
#
# Deconvoluting Peptide-MHCs...
# 100/100
# Deconvolution DONE.
#
# Reading alleles for Abelin...
# Reading alleles for Heyder...
# Reading alleles for Khoda...
#
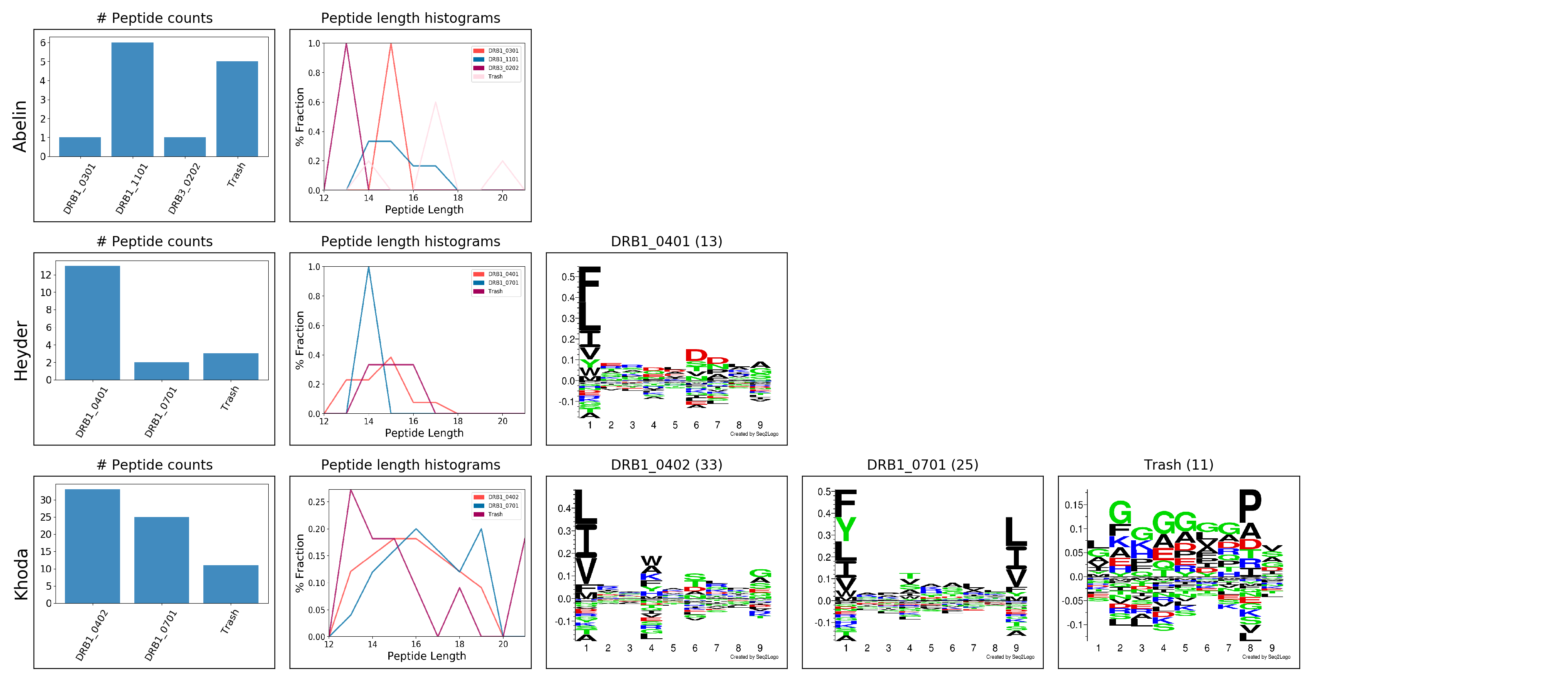
WARNING! Low quantity of sequences (1) found for DRB1_0301 in Abelin. This allele will not be plotted.
WARNING! Low quantity of sequences (6) found for DRB1_1101 in Abelin. This allele will not be plotted.
WARNING! Low quantity of sequences (1) found for DRB3_0202 in Abelin. This allele will not be plotted.
WARNING! Low quantity of sequences (5) found for Trash in Abelin. This allele will not be plotted.
#
# Found 13 sequences for DRB1_0401 in Heyder
WARNING! Low quantity of sequences (2) found for DRB1_0701 in Heyder. This allele will not be plotted.
WARNING! Low quantity of sequences (3) found for Trash in Heyder. This allele will not be plotted.
#
# Found 33 sequences for DRB1_0402 in Khoda
# Found 25 sequences for DRB1_0701 in Khoda
# Found 11 sequences for Trash in Khoda
#
# WARNING! The logo for DRB1_0301 in Abelin will not be generated.
# WARNING! The logo for DRB1_1101 in Abelin will not be generated.
# WARNING! The logo for DRB3_0202 in Abelin will not be generated.
# WARNING! The logo for Trash in Abelin will not be generated.
# 1/4 Making logo for DRB1_0401 in Heyder...
# WARNING! The logo for DRB1_0701 in Heyder will not be generated.
# WARNING! The logo for Trash in Heyder will not be generated.
# 2/4 Making logo for DRB1_0402 in Khoda...
# 3/4 Making logo for DRB1_0701 in Khoda...
# 4/4 Making logo for Trash in Khoda...
#
# Generating peptide count histograms... DONE.
#
# Generating peptide length histograms... DONE.
#
# Plotting /var/www/webface/tmp/server/mhcmotifdecon-1.0/61DCAF1F000013A1AED09789/run_21019/data/Abelin_count_histogram.png
# Plotting /var/www/webface/tmp/server/mhcmotifdecon-1.0/61DCAF1F000013A1AED09789/run_21019/data/Abelin_peptide_length_histogram.png
# Plotting /var/www/webface/tmp/server/mhcmotifdecon-1.0/61DCAF1F000013A1AED09789/run_21019/data/Heyder_count_histogram.png
# Plotting /var/www/webface/tmp/server/mhcmotifdecon-1.0/61DCAF1F000013A1AED09789/run_21019/data/Heyder_peptide_length_histogram.png
# Plotting /var/www/webface/tmp/server/mhcmotifdecon-1.0/61DCAF1F000013A1AED09789/run_21019/logos/Heyder@DRB1_0401.logo-001.png
# Plotting /var/www/webface/tmp/server/mhcmotifdecon-1.0/61DCAF1F000013A1AED09789/run_21019/data/Khoda_count_histogram.png
# Plotting /var/www/webface/tmp/server/mhcmotifdecon-1.0/61DCAF1F000013A1AED09789/run_21019/data/Khoda_peptide_length_histogram.png
# Plotting /var/www/webface/tmp/server/mhcmotifdecon-1.0/61DCAF1F000013A1AED09789/run_21019/logos/Khoda@DRB1_0402.logo-001.png
# Plotting /var/www/webface/tmp/server/mhcmotifdecon-1.0/61DCAF1F000013A1AED09789/run_21019/logos/Khoda@DRB1_0701.logo-001.png
# Plotting /var/www/webface/tmp/server/mhcmotifdecon-1.0/61DCAF1F000013A1AED09789/run_21019/logos/Khoda@Trash.logo-001.png
#
# SAVING LOGOS PLOT...
# DPI: 200.0
# PDF: /var/www/webface/tmp/server/mhcmotifdecon-1.0/61DCAF1F000013A1AED09789/run_21019/logos.pdf
# PNG: /var/www/webface/tmp/server/mhcmotifdecon-1.0/61DCAF1F000013A1AED09789/run_21019/logos.png
#
# MHC_Motif_Decon has finished.
#
# Results are stored in /var/www/webface/tmp/server/mhcmotifdecon-1.0/61DCAF1F000013A1AED09789/run_21019
###################################################
logos/
logos/Heyder@DRB1_0401.logo.eps
logos/Heyder@DRB1_0401.logo.txt
logos/Heyder@DRB1_0401.logo_freq.mat
logos/Heyder@DRB1_0401.logo-001.png
logos/Khoda@DRB1_0402.logo.eps
logos/Khoda@DRB1_0402.logo.txt
logos/Khoda@DRB1_0402.logo_freq.mat
logos/Khoda@DRB1_0402.logo-001.png
logos/Khoda@DRB1_0701.logo.eps
logos/Khoda@DRB1_0701.logo.txt
logos/Khoda@DRB1_0701.logo_freq.mat
logos/Khoda@DRB1_0701.logo-001.png
logos/Khoda@Trash.logo.eps
logos/Khoda@Trash.logo.txt
logos/Khoda@Trash.logo_freq.mat
logos/Khoda@Trash.logo-001.png
logos/Abelin_count_histogram.png
logos/Abelin_peptide_length_histogram.png
logos/Heyder_count_histogram.png
logos/Heyder_peptide_length_histogram.png
logos/Khoda_count_histogram.png
logos/Khoda_peptide_length_histogram.png
Motif Deconvolution plot:
Link to prediction file Output_file.xls
Link to tar.gz file with logo/image files data.tar.gz
Go
back.
ARTICLE ABSTRACTS
MAIN REFERENCE
Accurate MHC Motif Deconvolution of immunopeptidomics data reveals high relevant contribution of DRB3, 4 and 5 to the total DR Immunopeptidome
Saghar Kaabinejadian, Carolina Barra, Bruno Alvarez, Hooman Yari, William Hildebrand, Morten Nielsen
Mass spectrometry (MS) based immunopeptidomics is used in several biomedical applications including neo-epitope discovery in oncology, next-generation vaccine development and protein-drug immunogenicity assessment. Immunopeptidome data are highly complex given the expression of multiple HLA alleles on the cell membrane and presence of co-immunoprecipitated contaminants. The absence of tools that deal with these challenges effectively and guide the analysis and interpretation of this complex type of data is currently a major bottleneck for the large-scale application of this technique. To resolve this, we here present the MHCMotifDecon that benefits from state-of-the-art HLA class-I and class-II predictions to accurately deconvolute immunopeptidome datasets and assign individual ligands to the most likely HLA molecule, allowing to identify and characterize HLA binding motifs while discarding co-purified contaminants. We have benchmarked the tool against other state-of-the-art methods and illustrated its application on experimental datasets for HLA-DR demonstrating a previously underappreciated role for HLA-DRB3/4/5 molecules in defining HLA class II immune repertoires. With its ease of use, MHCMotifDecon can efficiently guide interpretation of immunopeptidome datasets, serving the discovery of novel T cell targets. MHCMotifDecon is available at https://services.healthtech.dtu.dk/service.php?MHCMotifDecon-1.0.
Frontiers in Immunology 26 January 2022.
Sec. Antigen Presenting Cell Biology, DOI: 10.3389/fimmu.2022.835454
Full text
EARLIER REFERENCES