The BepiPred-3.0 server predicts both linear and discontinous B-cell epitopes from protein sequence, using neural networks trained on state of the art protein language embeddings of epitope and non-epitope amino acids determined from crystal structures. A sequential smoothing (rolling mean) can be optionally used.
Inputs
The BepiPred-3.0 server requires protein sequence(s) in fasta format, and can not handle nucleic acid sequences.
- Protein sequence(s)
Paste protein sequence(s) in fasta format into the field. You can also load an example sequence, by clicking 'Load Data'.
You may also upload a fasta formatted file with protein sequence(s).
- Top epitope percentage cutoff
Specify a percentage cutoff a B-cell epitope predictions (20%, 50%, 70% or all %'s). The outputs are top % most likely B-cell epitope predictions.
- Threshold
A second output file is a fasta formatted containing B-cell epitope predictions at the set classification threshold.
You can specify classification threshold here (default is 0.1512)
- Sequential smoothing on graphical output (yes/no)
A third output file is a .html file with a graphical display of B-cell epitope predictions on the protein sequence(s).
You may specify to use a sequential smoothing (rolling mean) that makes it easier to identify patches of linear B-cell epitopes.
But the resulting predictions have a higher risk of including false positive epitope residues (default is 'no').
Outputs
A total of 4 output file types are generated, where epitope and non-epitope residues are indicated with uppercase and lowercase letters respectively.
One of these is a .html file containing interactive plots, which can also be used directly on the result page.
- Interactive plot(s) (HTML)
A html file containing the graphical outputs for B-cell epitope predictions.
The optimal threshold is often protein specific.
These figures allows a user manually set the threshold for each and get the corresponding B-cell epitope predictions.
- Raw output (CSV)
A csv file containing the B-cell epitope probability scores for each residue of the protein sequence(s).
The sequentially smoothed (linear epitope scores) are also provided.
- B Cell epitope predictions (FASTA)
A fasta file of the B-cell epitope predictions for the protein sequence(s) at the specified threshold.
B-cell epitope residues are indicated with uppercase.
- Top % epitope candidates (FASTA)
Fasta files with the percentwise most likely B-cell epitopes predictions for the protein sequence(s). This output includes prediction based on BepiPred-3.0 scoring as well as BepiPred-3.0 scoring with sequential smoothing (liner epitope scoring).
B-cell epitope residues are indicated with uppercase.
Graphical output
In the graphical output, B-cell epitope predictions are illustrated with bar plots.
The threshold for predicting B-cell epitopes is often protein-specific,
and single threshold is unlikey to be optimal for all proteins.
We believe this intuitive interface allows researchers to maximize their precision of B-cell epitope prediction.
Graph output without sequential smoothing (discontinous B-cell epitope prediction)
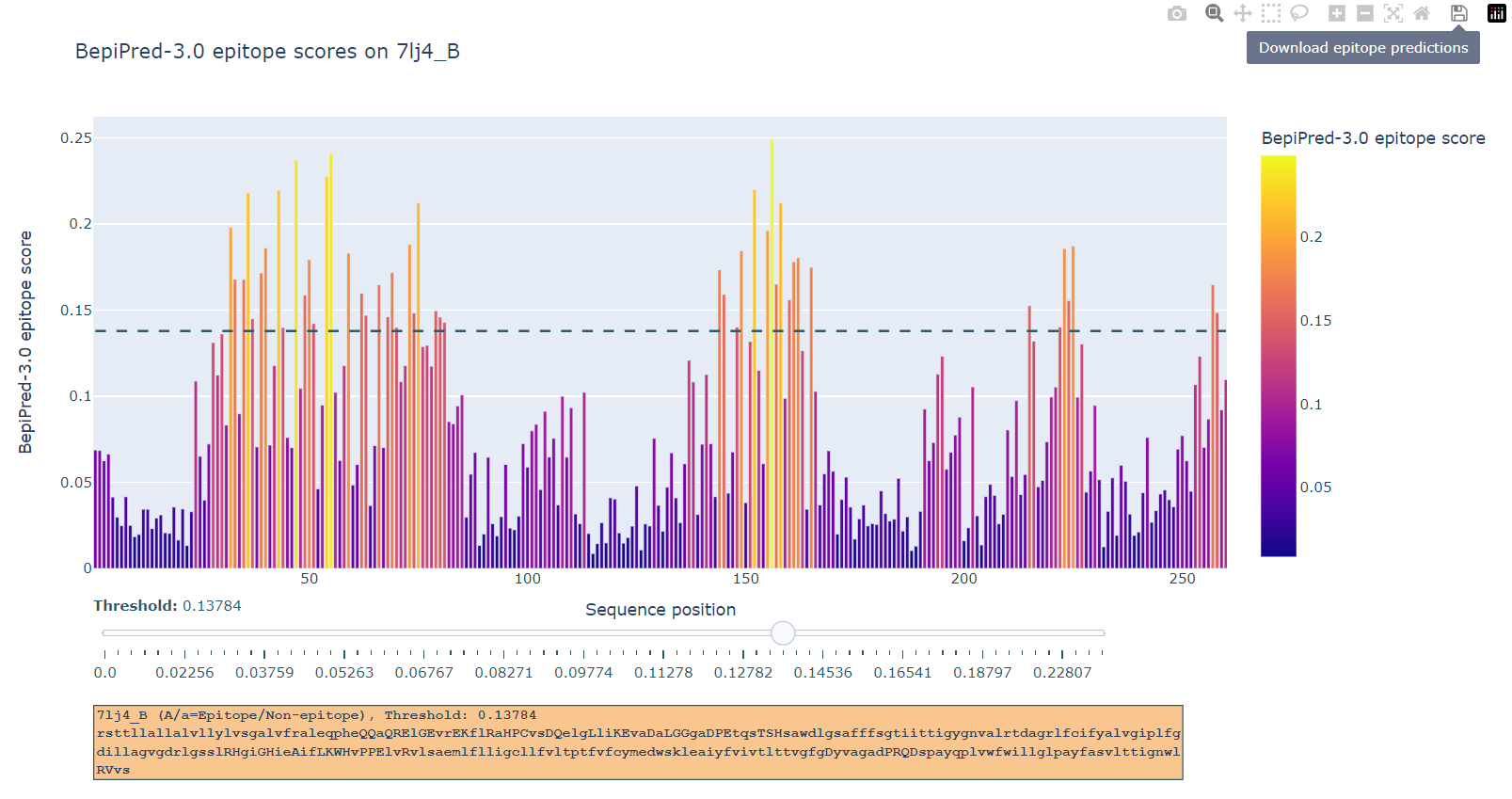
The x and y axis are protein sequence positions and BepiPred-3.0 epitope scores.
Residues with a higher score are more likely to be part of a B-cell epitope.
The threshold can be set by using the slider, which moves a dashed line along the y-axis.
Epitope predictions are updated according to the slider.
The B-cell epitope predictions at the set threshold can be downloaded by clicking the button 'Download epitope prediction'.
Graph output with sequential smoothing (linear B-cell epitope prediction)
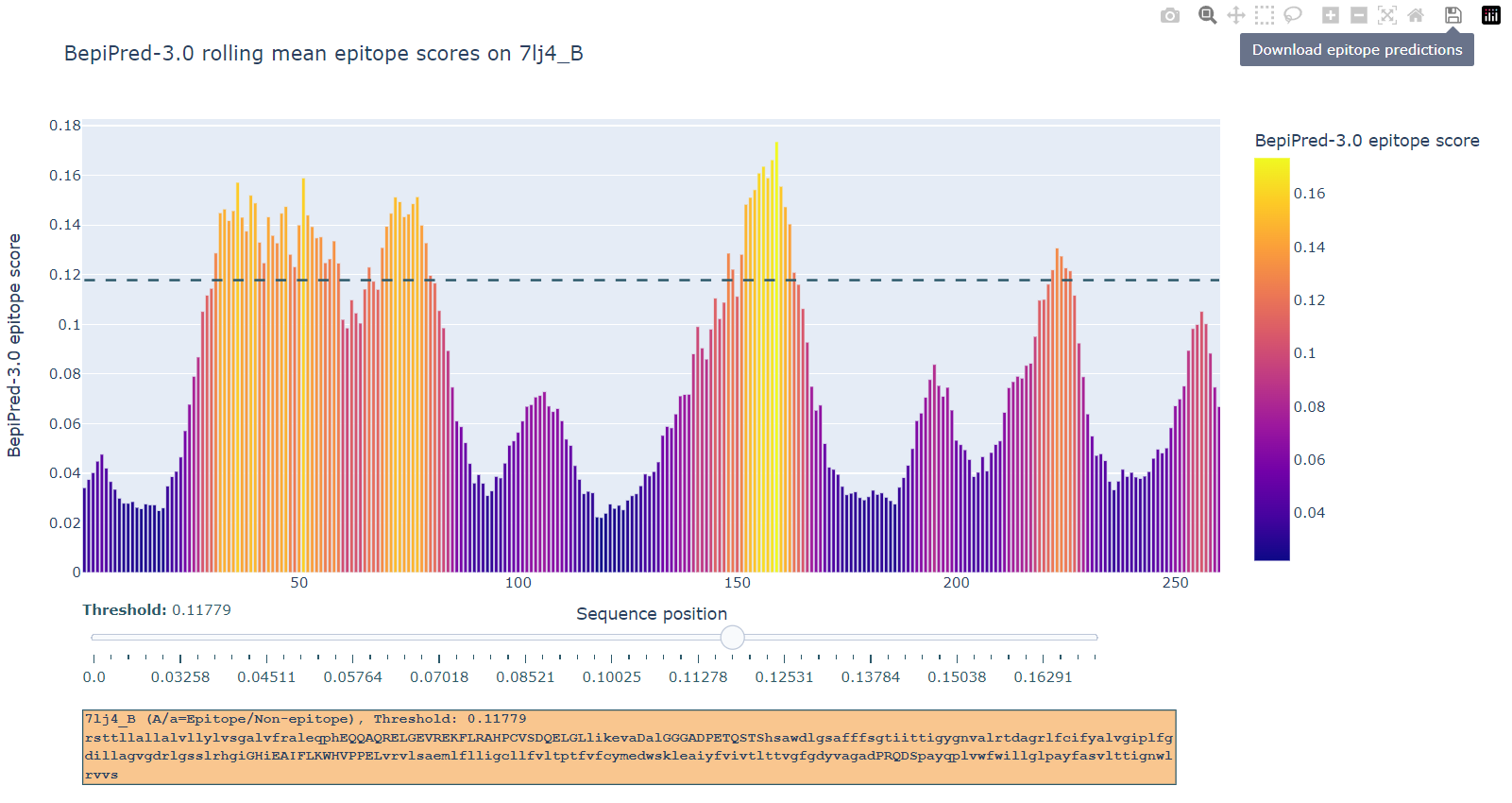
If you chose to use the sequential smoothing (rolling mean) option, the graphical output will look different.
Using this option is more useful for detecting linear epitopes.
But it is important to note, that some residues in the predicted linear epitope are false positives,
meaning that they do not interact directly with an antibody.
This is because BepiPred-3.0 is trained on PDB crystal structures of ab-ag complexes,
and to predict antigen residues that are in contact with an antibody (within 4 angstrom).